第六节 脂蛋白代谢紊乱
脂蛋白代谢紊乱的常见现象是血中Ch或TG升高,或者是各种脂蛋白水平异常增高。
高脂蛋白血症(hyperlipoproteinemia)是指血浆中CM、VLDL、LDL、HDL等脂蛋白有一类或几类浓度过高的现象。一般根据血浆(血清)外观、血总胆固醇、甘油三酯浓度以及血清脂蛋白含量将高脂蛋白血症进行分型。1967年Fredrickson将高脂血症分为六型,从生化及遗传角度考虑,有其不合理的一面,因为同一型高脂血症可有几种不同的基因型,临床实践表明,这种分型仍有一定的应用价值,因此,一直沿用至今。确认各种类型的高脂蛋白血症时,一定要用血清总胆固醇及甘油三酯、脂蛋白水平交叉检验,以求准确定型。因为任何一种高脂蛋白血症都有Ch、TG一项或两项指标同时升高即高脂血症(hyperlipidemia)出现。高脂血症与高脂蛋白血症实际上是同义词,而后者说明脂质代谢的病理变化更为确切。
从脂蛋白代谢紊乱的原因分类可分为原发性和继发性两大类。原发性是遗传缺陷所致,如家族性高胆固醇血症。继发性是继发于许多疾病所致,如糖尿病、肾病等疾患可继发引起高脂血症。下面着重介绍原发性高脂蛋白血症。
从遗传基因角度考虑,原发性高脂蛋白血症一定由遗传基因突变引起。从生化角度考虑是基因突变所致的基因表达的产物蛋白质水平上的缺陷,如Apo、酶和受体蛋白的异常。Apo异常多见于ApoE变异,典型例子是Ⅲ型高脂蛋白血症,ApoB变异可引起无β-脂蛋白血症,表现为脂肪吸收障碍;ApoAⅠ异常,则有Tangier病出现。导致血清HDL和ApoAⅠ水平降低;LCAT和LPL缺陷以及受体缺陷同样导致脂蛋白代谢异常,如家族性高Ch血症。
一、高脂蛋白血症
1967年Fredrickson等用改进的纸上电泳法分离血浆脂蛋白,将高脂蛋白症分为五型,即Ⅰ、Ⅱ、Ⅲ、Ⅳ和Ⅴ型。1970年世界卫生组织(WHO)以临床检验表现型为基础分为六型,将原来的Ⅱ型又分为Ⅱa和Ⅱb两型,如表4-1所示。
(一)Ⅰ型高脂蛋白血症
患者血浆TG升高,Ch正常,CM、VLDL含量升高,LDL及HDL均降低。约有2/3的病人在10岁前发病。
患者新鲜血清外观呈乳白色混浊,4℃过夜,血浆上层出现“奶油样”上层。大部分患者伴有视网膜脂血症、急性胰腺炎及肝脾肿大。
表4-1 人高脂蛋白血症分型及其特征
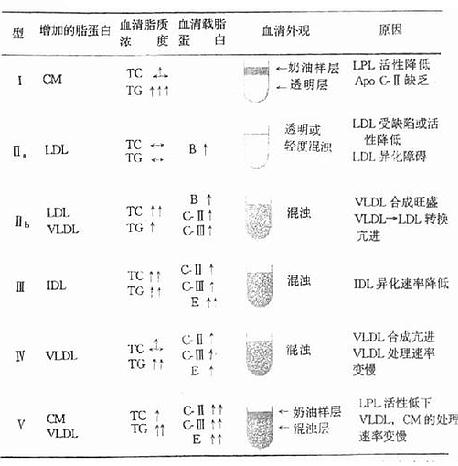
本症为常染色体隐性遗传,家族性遗传性纯合子型患者除血脂改变外,临床症状明显,而杂合子除TG高外,其症状不明显。Ⅰ型高脂血症又称为家族性高CM血症。发病原因主要是LPL的ApoCⅡ的遗传性缺陷,使LPL缺乏或者不能激活,CM中TG不能水解转变成CM残粒,无法被LDL受体识别进行代谢,从而造成CM在血浆中堆积。
(二)Ⅱa型高脂蛋白血症
Ⅱa型又称为家族性高胆固醇血症,血浆LDL和Ch明显升高。血清脂蛋白电泳呈现浓染的β-脂蛋白带,提示β-脂蛋白含量升高,故又称为高β-脂蛋白血症。纯合子病人在青春期即因动脉粥样硬化而死亡,这类病人除冠状动脉硬化外,还会出现黄色瘤和角膜弓状云等。
Ⅱa型有明显家族史,由LDL受体缺陷引起。纯合子型患者LDL受体完全缺陷,杂合子型者LDL受体只为正常的1/2。细胞不能通过膜上LDL受体从血中摄取LDL,使血浆中LDL升高。近年,Goldstein和Brown用纤维母细胞对遗传性高Ch血症遗传分析发现,LDL受体缺陷有三种不同的细胞表型:①无LDL受体型;②LDL受体缺陷型,细胞表面受体活性为正常的5%-20%;③入胞缺陷(内吞缺陷)型,即LDL受体可以与LDL结合,但是不能以正常速度内吞,从而导致LDL堆积于血浆中。目前所知,Ⅱa型是因为编码LDL受体的基因突变所致。
(三)Ⅱb型高脂蛋白血症
Ⅱb型又称为高β-脂蛋白及高前β-脂蛋白血症。血浆电泳图谱中,除β-脂蛋白增高外,前β-脂蛋白含量也升高,但二者并不融合。血浆中除LDL和Ch升高外,VLDL也升高,当然TG也升高,所以又称为混合型高脂蛋白血症。Ⅱb型与Ⅱa型的主要区别是前者LDL受体活性正常,患者多合并肥胖,糖代谢及胰岛素分泌异常,易伴发黄色瘤及动脉粥样硬化症。
Ⅱb型为显性遗传性疾患,体内VLDL合成量过多,ApoB100合成量比正常高两倍,LDL也增高。另外VLDL合成增加的同时,VLDL代谢分解速度并未增强,从而使过量合成的VLDL不能加速分解,造成血浆中VLDL蓄积,同时LDL代谢速度也减慢。
(四)Ⅲ型高脂蛋白血症
Ⅲ型属于家族遗传性高脂蛋白血症,血清电泳图谱上β-脂蛋白带与前β-脂蛋白带融合,呈现一个宽而浓染的色带,称为“阔β带”,因而出现β-移动度的VLDL,故也称为高β-VLDL血症。病因为ApoE异常,属显性遗传。正常人ApoE约65%-75%为E3/E3型,患者亦多见ApoE2/E2型,后者与受体结合力仅为前者的20%-40%。临床症状常见冠状动脉粥样硬化、外周血管病和黄色瘤等。
Ⅲ型患者代谢性改变是:①β-VLDL经肝的ApoE受体清除受阻;②肝从CM残粒获得的外源性胆固醇减少,自身合成Ch并分泌VLDL增多,使VLDL过度生成而堆积于血浆中;③LPL活性降低,VLDL在体外不能转变成LDL。Ⅲ型患者除β-VLDL出现外,Ch和TG均升高。Ⅲ型比较罕见。
(五)Ⅳ型高脂蛋白血症
Ⅳ型又称为家族性高甘油三酯血症或高VLDL血症,仅血浆TG升高,LDL正常,HDL降低。Ⅳ型高TG症的患者属于常染色体显性遗传。Ⅳ型发生动脉粥样硬化的危险性也增加,但不如Ⅱ型、Ⅲ型严重。发病原因尚不清楚。
(六)Ⅴ型高脂蛋白血症
Ⅴ型患者血清脂蛋白电泳图谱呈现CM及前β-脂蛋白深染,属于高CM和高前β-脂蛋白血症都存在的混合型高脂蛋白血症,故又称为高CM与高前β-脂蛋白血症。将该型血浆置于4℃冷藏10小时,可见上层为“奶油样”,下层为混浊状,属于罕见的血清外观。血清TG远远超出正常,LDC-C和HDL-C低于正常值,Ch在正常范围,VLDL-C超出正常范围,VLDL-C/VVLDL-TG低于0.3。若给予患者注入肝素后,血清CM消失。与Ⅲ型不同点是,Ⅴ型人血清VLDL-C/VLDL-TG为0.3以上。该型患者,若仅是LPL活性降低属显性遗传;如既有LPL活性降低,又有ApoCⅡ减少,则属于隐性遗传。常于20岁前发病,家族史明显的Ⅴ型者,丘疹状黄色瘤的发病率可高达30%-50%,并伴有急性胰腺炎及肝脾肿大。ApoCⅡ缺乏、LPL活性降低、VLDL生成过多而代谢率降低等原因导致VLDL在血内堆积。
各类型的高脂蛋白血症表型的诊断指标的变化值如表4-1所示
(七)高HDL血症
血浆HDL含量过高导致高HDL血症,也属于病理状态。HDL具有抗动脉粥样硬化作用,是人们公认的,然而并非血浆HDL含量越高越好。血浆HDL-胆固醇(HDL-C)含量超过1g/L,定义为高HDL血症。现已查明,高HDL血症是因为有CETP和HTGL等活性异常所致。高HDL血症分为原发性和继发性。原发性高HDL血症的病因有以下几种可能:①CETP缺损;②HTGL活性降低;③其他不明原因。继发性高HDL血症病因有:①运动失调;②饮酒过量;③原发性胆汁性肝硬化;④治疗高脂血症的药物引起;⑤其他原因。总之,CETP及HTGL活性降低是引起高HDL血症的主要原因。若CETP缺陷,HDL上的CE蓄积,使HDL增多;若HTGL活性降低,HTGL与HDL被肝细胞摄取减少并使HDL2→HDL3转换过程减慢而停留在血液中,并使其浓度增加,出现高HDL血症。血清中总胆固醇轻度或中度升高,HDL-C高达正常人3-5倍,血清ApoAⅠ、CⅢ、E明显增加,ApoB呈低值。因为CETP活性低,从HDL转运到含ApoB的脂蛋白的CE量减少,即运输障碍使CE量增加。高HDL血症多见于CETP缺乏者,易出现多分散LDL,而HDL颗粒变大。有实验表明,CETP活性低的动物作胆固醇负荷实验很容易形成动脉粥样硬化,对人而言,高HDL血症与动脉粥样硬化的关系有待进一步研究。
HDL按超速离心法可分为HDL2和HDL3。用聚丙烯酰胺梯度凝胶电泳可将HDL分为HDl1、2a、2b、3a、3b、3c六种亚组份颗粒,如图4-10所示。有报道冠心病患者大型颗粒HDL2b减少,小颗粒HDL3b增加,认为HDL2b的上升有抗动脉粥样硬化的作用,HDL3b颗粒的增加则可以引起脂质代谢障碍,动脉粥样硬化形成。
高脂蛋白血症分为六型,在临床诊治疾病过程中有一定的意义。从临床实验室诊断方法学考虑,作脂蛋白检测有一定难度,电泳分离法欠准确,按超速离心法,操作时间过长,难以快速定量。另外,高脂蛋白血症多数与遗传有关。目前可采用载脂蛋白基因分型以弥补按脂蛋白进行分型的不足之处。载脂蛋白的基因分型是目前研究脂质代谢及其探讨动脉粥样硬化发病机制的热门话题。
二、遗传性脂蛋白代谢异常
(一)ApoAⅠ异常症
Assmann分析近两万人,发现每500人中有1例ApoAⅠ结构基因杂合子出现,比野生型多一个或少一个正电荷或负电荷。大多数变异无明显血脂的变化。仅有ApoAⅠMarburg病在107位上的Lys缺失,引起轻度的TG升高。ApoAⅠ的Milano变异体(173Arg→Cys)血浆中HDL有所降低,然而冠心病发病率未见增加。ApoAⅠ和ApoCⅢ基因重排导致的变异可引起家族性ApoAⅠ和ApoCⅢ缺乏症,用EcroⅠ限制性内切酶分析ApoAⅠ基因,发现家族性早发性冠心病患者都出现6.5kb片段纯合子,正常人为13kb纯合子,其杂合子为13kb/6.5kb,推测纯合子6.5kb与动脉粥样硬化发病有关。
ApoAⅠ与ApoCⅢ缺陷者表现为血HDL水平降低,易出现早期动脉粥样硬化。有报道ApoAⅠ减少会导致LCAT活性降低,使含ApoCⅠ、ApoAⅢ的脂蛋白如CM置换发生障碍,从而在体内蓄积。
(二)ApoB异常症
ApoB缺陷将出现无β-脂蛋白血症或低β-脂蛋白血症。无β-脂蛋白血症是纯合子隐性遗传病,称为Bassen-Kornzeig综合征,有脂肪吸收障碍(脂肪泻)、红细胞变形(棘状红细胞症)和运动失调等症状。
低β-脂蛋白血症为显性遗传病,杂合子者血中LDL浓度低,与无β-脂蛋白血症有区别。经三个家族分析,患者肠粘膜细胞的ApoB48合成正常而不能合成ApoB100,即ApoB48外显子以外的ApoB100外显子领区异常,由于LDL受体领域附近的点突变(Arg3500→Glu),使LDL受体结合能力降低。
ApoB100的羧基端是其与LDL受体的结合区,有下列依据证明这一推测:①观察到30种单克隆抗体能与LDL受体结合的肽链区域结合,即ApoB100的2980-3780一段可被阻止,其他单克隆抗体无阻止作用;②缩短的ApoB异构体缺乏羧基端,则不能与LDL受体结合;③羧基端有三个区域可与肝素结合,该区域含有几簇带正电荷的氨基酸残基,其中之一的序列与ApoE区域的序列有同源性,这一区域称为ApoE区,已证实这一区域可与LDL受体结合,其序列为3359-3367氨基酸残基位置,带正电荷的氨基酸簇相当保守;④经化学修饰的带正电荷的ApoB100不能与HDL受体结合;⑤若ApoB100分子中Arg→Gln3500,LDL分子不能与LDL受体结合,导致高脂蛋白血症,如Innerarity的家族性高脂蛋白血症。
ApoB100的cDNA中某一核苷酸的变异或缺失,均可引起家族性低β-脂蛋白血症,迄今已发现有25种之多,血浆LDL浓度降低,杂合子患者血浆中ApoB和LDL浓度为正常的1/4-1/2。一般无症状,纯合子则更为严重,包括脂肪吸收不良、棘型红细胞、视网膜色素沉着和神经性肌肉退变。
ApoB100在血浆脂蛋白中分子量最大,氨基酸链最长,因此在合成蛋白质和形成脂蛋白的过程中,任何部位或环节均可能发生变异,可想而知,今后发现的ApoB100的变异会更多。
(三)ApoCⅡ异常症(遗传变异)
ApoCⅡ缺陷导致LPL活性降低。因为ApoCⅡ是LPL发挥催化作用不可缺少的辅因子。ApoCⅡ异常会出现高TG血症,高CM血症,发病率约1/10万。现已有ApoCⅡ有多种变异体的报道。
(四)ApoE异常症
ApoE是LDL受体的配体,其表型不同,与LDL受体结合的能力也不同,E4和E3几乎相同,E2几乎无结合功能。E2纯合子因为第158氨基酸残基突变,CM残粒或β-VLDL滞留导致高Ch、TG血症,高脂蛋白血症,易出现早期动脉粥样硬化。典型例子是家族性Ⅲ型高脂血症,ε2基因纯合子人群分布频率为1%,家族性Ⅲ型高脂血症发病率为10000人有2-3人。究其病因,ApoE2纯合子遗传缺陷因素是主要的,然而还有环境及生理性因素等的影响,如甲状腺功能亢进、肿瘤以及家族性复合型高脂蛋白血症等。
(五)LDL受体异常
LDL受体异常导致FH发生,属显性遗传,遗传频率约1/500。杂合子的高LDL血症易导致动脉粥样硬化。FH的LDL受体基因变异和LDL受体合成的过程中均可出现异常,将其分为四类:①单元1异常是因为mRNA转录障碍导致总体蛋白性质改变,生物学活性降低;②单元2异常是分子量为12万的受体前驱体异常,从小胞体到高尔基复合体运送障碍,ER潴留,与单元2相同,富含Cys领域阅读框的缺失(in-framedeletion)存在;③单元3变异,细胞表面的分子量160ku的成熟受体数量显著减少,使LDL受体结合能力下降;④单元4变异为受体不能局部化,使LDL无法结合而进入细胞内。这几种变化均与lDL受体结构有关。
(六)LPL与HTGL异常症
LPL与ApoCⅡ异常同样都是出现高CM血症,而血中VLDL并不升高,常伴有胰腺炎产生。
HTGL缺乏,有与Ⅲ型高脂血症类似的症状,CM残粒滞留。
(七)LCAT异常症
LCAT缺乏者,HDL中CE比例增加,使HDL处于新生未成熟圆盘状态,相反LDL的CE减少,TG增多。有角膜混浊、肾损害、溶血性贫血等症状,鱼眼病就是LCAT基因突变,使Cys替代Arg引起LCAT活性降低,致使HDL结构变化,并使血浆中ApoAⅠ、Ⅱ和HDL浓度只有正常人的20%。
(八)CETP异常症
CETP缺陷者或者活性受到强烈抑制则呈现高HDL血症,血浆LDL浓度降低,同时还有可能出现动脉粥样硬化症。
三、继发性高脂蛋白血症
某些原发性疾病在发病过程中导致脂质代谢紊乱,进而出现高脂蛋白血症,称为继发性高脂蛋白血症,引起继发性高脂血症或高脂蛋白血症的病因是多方面的,如糖尿病、肾病及某些内分泌紊乱等疾患。
99-12-13 9:24 刘小琴 校对
, http://www.100md.com