遗传性球形细胞增多症
http://www.100md.com
大众医药网
【概述】
遗传性球形细胞增多症(hereditary spherocytosis,HS),是一种红细胞膜缺陷的溶血性贫血。临床主要特征有球形细胞显著增多,对低渗盐液脆性增加,并有不同程度的黄疸和脾肿大。脾切除疗效良好。在遗传性溶血性贫血中发病率最高。
【病因】
本症患者的红细胞膜结构异常是由不同的膜支架异常所引起。部份患者有膜的收缩蛋白缺乏,对蛋白水解酶极为敏感,可将变性的膜收缩蛋白降解为小肽,从红细胞膜上丢失,使膜表面积减少而变为球形。膜收缩蛋白与蛋白区带4.1结合能力减少30%左右,致使膜收缩蛋白与肌动蛋白结合能力明显削弱。红细胞蛋白质激酶缺乏,使收缩蛋白的磷酸化减弱,影响变形性能。
由于原发性膜缺陷,膜的被动性钠盐流入的通透性增加,水随钠盐而进入细胞内,使凹盘形细胞表面积减少,逐渐变小而厚,接近于球形。为了保持细胞内外钠盐浓度的正常比例,就需要产生更多的三磷酸腺苷(ATP),以加速钠的排出和钾的摄入。所以球形细胞的糖酵解率往往较正常红细胞增加20%~30%,以补偿大量ATP的消耗。ATP的相对缺乏使膜上钙-活性ATP酶受到抑制,钙容易沉积在膜上。胞膜中肌动凝蛋白(actomyosin)由溶胶变为凝胶,因而红细胞膜变僵硬,丧失柔韧性。球形细胞的直径虽然小于6μ左右,但由于细胞膜变形性和柔韧性减退而被阻留在脾索内,不能通过内皮细胞间空隙(直径仅为3μ左右)进入脾窦。大量红细胞在脾索内滞留过程中,ATP及葡萄糖进一步消耗,代谢缺陷更形加剧,终至破坏而溶解。
, http://www.100md.com
【临床表现】
本症大部分为常染色体显性遗传,极少数为常染色体隐性型。男女均可发病。常染色体显性型特征为贫血、黄疸及脾肿大。根据疾病严重度分为以下三种:①轻型多见于儿童,约占全部病例的1/4,由于骨髓代偿功能好,可无或仅有轻度贫血及脾肿大;②中间型约占全部病例2/3,多成年发病,有轻及中度贫血及脾肿大;③重型仅少数患者,贫血严重,常依赖输血,生长迟缓,面部骨结构改变类似海洋性贫血,偶尔或一年内数次出现溶血性或再生障碍性危象。常染色体隐性遗传者也多有显著贫血及巨脾,频发黄疸。溶血或再障危象常因感染、妊娠或情绪激动而诱发,患者寒战、高热,恶心呕吐,急剧贫血,持续几天或甚至1~2周。本症患者较多见(约有50%)的并发症是由于胆红素排泄过多,在胆道内沉淀而产生胆石症,其次是发生于踝以上的腿部慢性溃疡,常迁延不愈,但可经脾切除而获得痊愈。发育异常或智力迟钝很罕见。
【实验检查】
除非有急性发作,贫血一般不重,但危象时血红蛋白可低至3g/dl左右。部分红细胞(20%~30%)直径较小,但比正常厚,在涂片中显得小而染色深,所以MCV轻度减少,MCHC增多。网织红细胞经常在5%~20%之间,而在急性溶血发作后可高达0~70%,血中伴有少数幼红细胞。在低渗盐液中红细胞的渗透性脆性,随球形红细胞的增多而增强。脆性试验的曲线形态不一。当有相当数量的红细胞呈球形时,大部分曲线移向正常曲线之右。如球形细胞不多,脆性试验曲线仍可在正常范围,但其尾端则在较高浓度的盐水中。将患者红细胞孵育24小时后,再进行脆性试验,即使极轻度患者,也可显示渗透性脆性增加(图20-8)。自体溶血试验阳性,待加葡萄糖后可予纠正。骨髓多呈正常幼红细胞增生象。血清总胆红素在17.1~68.4μmol/L。当再生障碍危象发生时;红细胞数急剧下降,但网织红细胞反而消失。血清总胆红素不一定增加而反减少。骨髓内幼红细胞生成不良或甚至成熟停顿。少数病例可伴有白细胞及血小板减少。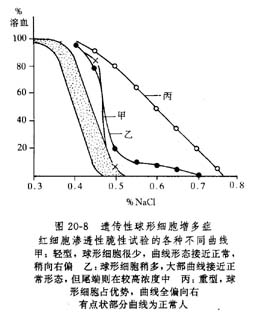
, 百拇医药
【诊断说明】
典型病例具有脾大、黄疸、贫血、球形细胞增多与红细胞渗透脆性增加,诊断一般不难。家族调查可帮助确立诊断。红细胞孵育后再进行脆性试验,有助于发现轻型病例。血片中球形细胞不多的病例,应与自身免疫性溶血性相鉴别。
【治疗说明】
脾切除对本病有显著疗效。持续多年的黄疸和贫血在手术后大都很快消失,但一定程度的球形细胞依然存在,红细胞渗透性脆性仍然增高。症状较重或屡有急性发作;症状在幼年期出现,即使病情较轻,为防止日后急性发作或并发症起见,都应考虑手术治疗。在儿童期确立诊断,如无特殊必要,手术宜在6岁后进行较为妥当。中年以上患者如症状轻微,仅注意一般卫生保健和适当的生活制度即可。输血疗效并不持久,仅留作术前准备和危象时应用。, http://www.100md.com
遗传性球形细胞增多症(hereditary spherocytosis,HS),是一种红细胞膜缺陷的溶血性贫血。临床主要特征有球形细胞显著增多,对低渗盐液脆性增加,并有不同程度的黄疸和脾肿大。脾切除疗效良好。在遗传性溶血性贫血中发病率最高。
【病因】
本症患者的红细胞膜结构异常是由不同的膜支架异常所引起。部份患者有膜的收缩蛋白缺乏,对蛋白水解酶极为敏感,可将变性的膜收缩蛋白降解为小肽,从红细胞膜上丢失,使膜表面积减少而变为球形。膜收缩蛋白与蛋白区带4.1结合能力减少30%左右,致使膜收缩蛋白与肌动蛋白结合能力明显削弱。红细胞蛋白质激酶缺乏,使收缩蛋白的磷酸化减弱,影响变形性能。
由于原发性膜缺陷,膜的被动性钠盐流入的通透性增加,水随钠盐而进入细胞内,使凹盘形细胞表面积减少,逐渐变小而厚,接近于球形。为了保持细胞内外钠盐浓度的正常比例,就需要产生更多的三磷酸腺苷(ATP),以加速钠的排出和钾的摄入。所以球形细胞的糖酵解率往往较正常红细胞增加20%~30%,以补偿大量ATP的消耗。ATP的相对缺乏使膜上钙-活性ATP酶受到抑制,钙容易沉积在膜上。胞膜中肌动凝蛋白(actomyosin)由溶胶变为凝胶,因而红细胞膜变僵硬,丧失柔韧性。球形细胞的直径虽然小于6μ左右,但由于细胞膜变形性和柔韧性减退而被阻留在脾索内,不能通过内皮细胞间空隙(直径仅为3μ左右)进入脾窦。大量红细胞在脾索内滞留过程中,ATP及葡萄糖进一步消耗,代谢缺陷更形加剧,终至破坏而溶解。
, http://www.100md.com
【临床表现】
本症大部分为常染色体显性遗传,极少数为常染色体隐性型。男女均可发病。常染色体显性型特征为贫血、黄疸及脾肿大。根据疾病严重度分为以下三种:①轻型多见于儿童,约占全部病例的1/4,由于骨髓代偿功能好,可无或仅有轻度贫血及脾肿大;②中间型约占全部病例2/3,多成年发病,有轻及中度贫血及脾肿大;③重型仅少数患者,贫血严重,常依赖输血,生长迟缓,面部骨结构改变类似海洋性贫血,偶尔或一年内数次出现溶血性或再生障碍性危象。常染色体隐性遗传者也多有显著贫血及巨脾,频发黄疸。溶血或再障危象常因感染、妊娠或情绪激动而诱发,患者寒战、高热,恶心呕吐,急剧贫血,持续几天或甚至1~2周。本症患者较多见(约有50%)的并发症是由于胆红素排泄过多,在胆道内沉淀而产生胆石症,其次是发生于踝以上的腿部慢性溃疡,常迁延不愈,但可经脾切除而获得痊愈。发育异常或智力迟钝很罕见。
【实验检查】
除非有急性发作,贫血一般不重,但危象时血红蛋白可低至3g/dl左右。部分红细胞(20%~30%)直径较小,但比正常厚,在涂片中显得小而染色深,所以MCV轻度减少,MCHC增多。网织红细胞经常在5%~20%之间,而在急性溶血发作后可高达0~70%,血中伴有少数幼红细胞。在低渗盐液中红细胞的渗透性脆性,随球形红细胞的增多而增强。脆性试验的曲线形态不一。当有相当数量的红细胞呈球形时,大部分曲线移向正常曲线之右。如球形细胞不多,脆性试验曲线仍可在正常范围,但其尾端则在较高浓度的盐水中。将患者红细胞孵育24小时后,再进行脆性试验,即使极轻度患者,也可显示渗透性脆性增加(图20-8)。自体溶血试验阳性,待加葡萄糖后可予纠正。骨髓多呈正常幼红细胞增生象。血清总胆红素在17.1~68.4μmol/L。当再生障碍危象发生时;红细胞数急剧下降,但网织红细胞反而消失。血清总胆红素不一定增加而反减少。骨髓内幼红细胞生成不良或甚至成熟停顿。少数病例可伴有白细胞及血小板减少。
, 百拇医药
【诊断说明】
典型病例具有脾大、黄疸、贫血、球形细胞增多与红细胞渗透脆性增加,诊断一般不难。家族调查可帮助确立诊断。红细胞孵育后再进行脆性试验,有助于发现轻型病例。血片中球形细胞不多的病例,应与自身免疫性溶血性相鉴别。
【治疗说明】
脾切除对本病有显著疗效。持续多年的黄疸和贫血在手术后大都很快消失,但一定程度的球形细胞依然存在,红细胞渗透性脆性仍然增高。症状较重或屡有急性发作;症状在幼年期出现,即使病情较轻,为防止日后急性发作或并发症起见,都应考虑手术治疗。在儿童期确立诊断,如无特殊必要,手术宜在6岁后进行较为妥当。中年以上患者如症状轻微,仅注意一般卫生保健和适当的生活制度即可。输血疗效并不持久,仅留作术前准备和危象时应用。, http://www.100md.com