代谢是人体从所吃食品的脂肪、碳水化合物、和蛋白质中获得能量并合成所需分子的手段,代谢也是通过矿物质和维生素帮助酶反应的手段。
综合状态使发生在细胞中的酶催化反应变成复杂的网络。虽然本报告专指由代谢过程差错引起的疾病,但确实存在耐受系统差错的有效水平:一种酶突变经常地不是个体遭受疾病的原委。许多不同的酶可能完成修正同样的分子,可能有许多方发为各种代谢中间体获得最终结果。如果关键酶失效或者代谢机制途径受影响,只有此时会生病。
在这儿我们强调,对于基因已确认、克隆、和复制的代谢疾病,许多是代谢先天失调:由于是代谢酶突变的遗传因素,另一种是与调节蛋白和输送机制的突变有关。

第一节 BEST疾病
通常以卵磷脂斑营养不良症二型为特证的BEST疾病是主要发生在European Cancasians的一种遗传疾病。虽然受影响患者经常显示出症状而且这些症的严重性很不相同,但具有这类疾病的患者总是在他们的早年逐步显示出失去视敏度。
BEST疾病是一种常规染色体显性,换句话说,位于染色体11上VMD2基因的唯一复制发生突变可能引起疾病的发展。在他们失去视力前,具有这类疾病的患者在对应中心视觉的视网膜区域中积累了大量类似蛋黄(卵磷脂即是蛋黄的意思)物质。与其说打破了这些物质倒不如说形成与BEST疾病有关的逐渐丧失视觉的特征。
虽然已知VMD2基因功能局限于称为视网膜色素上皮的眼睛区域,但很少知道有关VMD2基因的蛋白质产物。根据VMD2蛋白质编码可能包括光受体成份的移去或加工的推测,VMD2蛋白质功能的检测和动物模型的开发将是朝着未来理解BEST疾病的决定性的步履。

第二节 肾上腺脑白质营养不良症
肾上腺脑白质营养不良症(ALD)是一种罕见的遗传性代谢疾病,它影响年青男孩的Lorenzo Odone,它是在1993年的影片“Lorenzo's oil”中报道的,在这种疾病中,失去了覆盖在脑神经上的脂肪(髓磷脂鞘),导致渐进性的神经功能丧失和死亡。
具有ALD疾病的患者,因为不会以正常的方式通过酶来分解脂肪酸,所以在他们的脑和肾上腺皮质中累积了高级的饱和、非常长键的脂肪酸。当ALD基因在1993年被发现后,人们惊叹是对应的蛋白质而不是酶,事实上是输送蛋白质家系中的成员,。关于输送蛋白质如何对脂肪酸酶的功能起作用,就这点而论,非常长链高级脂肪酸如何引起神经纤维上髓磷脂的失落仍是一个谜。直到最近在酵母菌酵母啤酒中曾找到所有有关ALD蛋白质的输送者后,才为人类疾病开发了白鼠模型。对白鼠模型的研究和其它分子生物学有进一步理解并加快有效治疗的程序。

第三节 戈谢病
戈谢病(Daucher disease发音“go-shay”)是一种由基因突变引起的遗传疾病。基因在正常情况下负责葡糖脑苷脂酶的作用,这种酶帮助人体分解成名为葡糖脑苷脂的脂肪。有戈谢病的患者,不能产生这种酶,因而也不能分解脂肪,这样就把脂肪累积在肝脏、脾和骨髓中。戈谢病能导致疼痛、疲劳、黄疸、骨损伤、贫血甚至死亡。
虽然戈谢疾病可能影响任何种族的人员,但对来自东欧(Ashkenazi)的犹太人影响更为普遍。在Ashkenazi犹太人口中,戈谢病是最普遍的遗传疾病,大约1/450人的概率。在总体人口中,影响的概率是1/10万,按照美国国家戈谢疾病基金的统计,有2500位美国人患戈谢病。
在1991年,酶取代治疗变成对戈谢病第一有效治疗方法,治疗由根据研究提供的改性型葡糖脑苷脂酶组成。治疗只要在门诊部进行。每隔两周治疗一次,每次花1—2小时。酶取代疗法能停止和经常逆转戈谢病的症状,让患者享受更好的生活质量。

第四节 葡萄糖、半乳糖吸收障碍
葡萄糖、半乳糖吸收障碍(GGM)是一种罕见的代谢障碍疾病,它是通过小肠内壁转化葡萄糖和半乳糖受障碍所引起的。GGM是以生命第一天的早期患严重腹泻和脱水为特征的,如果乳糖(奶糖)、蔗糖(片状糖)、葡萄糖、和半乳糖不能从膳食中移去,就会很快引起死亡。在世界上发现的200个严重GGM患者中,其中一半人是起因于近亲结婚。总人口中至少有10%的人表现为不耐葡萄糖,然而,这些人群只表现出轻微的疾病症状。
GGM是一种常规染色体隐性混乱,受此影响的个体在位于第22位染色体上遗传了两个SGLTI基因的缺陷拷贝。正常情况下,在小肠(称为腔)关闭空间,乳糖酶把乳糖分解成葡萄糖和半乳糖,而蔗糖酶把蔗糖分解成葡萄糖和果糖,由SGLT1的蛋白质产物然后把葡萄糖和半乳糖从小肠腔送至小肠细胞。而由GGM个体携带的突变产生失去功能的截短SGLTI1蛋白质,或出现不能转化小肠腔外葡萄糖和半乳糖的不适宜蛋白质,如果剩余的葡萄糖、半乳糖不能转化,身体脱水进入小肠腔就导致腹泻。
虽然不能治愈GGM疾病,但病人能通过在膳食中移去乳糖、蔗糖、和半乳糖的方法来控制他们的症状。出生前诊断为GGM的婴儿将付以以果糖为基准的取代公式,随后能否继续有正常的体格取决于以果糖为基准的固体膳食。具有严重GGM疾病的较大儿童和成年人也能采用以果糖为基准的膳食控制住他们的症状,并能显示出作为他们年令的改善葡萄糖允许量。

第五节 动脉粥样硬化
动脉粥样硬化,虽然年令到40或50岁才具有威胁性,但是一种能影响任何年令人群的疾病。它是一种以免疫系统细胞富胆固醇引起动脉狭窄为特征的疾病,它使能遗传或受环境影响的动脉粥样硬化的主要危险因素是:包括血液中胆固醇和三甘脂水平升高,高血压和吸烟。
能以几种不同形式存在的称为载脂蛋白E的蛋白质是一种在染色体19上找到的基因编码。它能从血液中移去过量的蛋固醇并将它带至肝细胞表面的受体上,一旦缺乏载脂蛋白E,就会失去携送到受体上去的能力,就会导致增加一个人的血脂升高,随后出现动脉硬化的危险。
当前,激烈争论的焦点是脂质蛋白E的各种突变形式如何影响人体。因此,许多被提出的建议停留在他们的实验阶段,而鼠正在证明能用于人类疾病模型,在我们充分理解脂质蛋白E在血液中脂质含量规律的机理以前,仍需要作大量的研究工作。

第六节 眼睛脉络膜的螺旋状萎缩
遭受脉络膜的螺旋状萎缩以及视网膜面临漸进性失明的人群,全盲通常发生在40至50岁的年令。该疾病是一种先天性代谢障碍。
在染色体10上,找到引起螺旋状萎缩的基因突变,编码一种称为鸟氨酸酮酸转氨酶(OAT)。不同的OAT遗传突变引起疾病症状的严重性不同。OAT把氨基酸鸟氨酸从尿环形式最后转化成谷氨酸盐。在螺旋状萎缩中,由于缺乏OAT功能,就增加了鸟氨酸的等离子水平。
现在知道在膳食中减少精氨酸有利于大多数病人,近来关于该疾病的研究包括:(1),研究等位基因(基因遗传的转位)相互作用的不同突变如何引起不同的疾病症状。(2),对鼠模型疾病的研究正在促使我们更好地理解疾病并希望达到治愈的目的。

第七节 糖尿病
糖尿病是一种影响人体制造和使用胰岛素的慢性疾病,激素必须把食物转化为能量。糖尿病很大程度上增加了视觉缺损、心脏病、肾脏衰竭、神经性疾病和其它疾病。在美国有160万受到I型糖尿病的影响,或青少年开始得糖尿病变更是严重的问题。
已经知道I型糖尿病具复杂的因素,即意味着有几种基因的突变影响疾病。例如,现在已经知道依赖胰岛素型糖尿病在位于第6条染色体上可能包含至少一个对I型糖尿病敏感的基因。虽然基因定位于还有抗原基因(正常产生免疫系统的分子不会攻其本生)的第六条染色体区域,但在这一位置上的突变如何真正地使病人发生危险还不清楚。
在I型糖尿病人中,人体免疫系统设置一种免疫,攻击它自身胰岛素和制造胰岛素的胰管细胞。然而,如何发生的机理还不清楚。
在人类基因组中,大约10个位点现在被发现似乎对I型糖尿病敏感,在这些中是:(1),在第11条染色体位点IDDM2上的基因。(2),
葡萄糖激酶基因,这是对帮助调节胰岛素分泌的葡萄糖代谢起关键作用的酶,它存在于第七条染色体上。
谨慎的病人保健和控制每日的胰岛素用量能较好地帮助病人获得健康。但为了防止常引起糖尿病的免疫应答,我们将需要用病鼠模型作实验,并进一步帮助我们理解在其它染色体上的基因如何增加病人的糖尿病危险性。

第八节 肥胖
肥胖是人体脂肪过量,它常损害健康。医生一般认为男子人体脂肪超过25%、女子人体脂肪超过30%就是肥胖。众所周知,论证肥胖是诸如心脏病、糖尿病、高血压、中风、某些癌的形成等慢性疾病的危险因素。论证证明肥胖形成有多种原因:基因、环境、生理、和其它因素都起作用。
由脂肪细胞产生的激素Leptin大约三年前在鼠中发现的。随后人类Ob基因被定位于第七条染色体上,当大量脂肪储藏在脂肪细胞中时,Leptin起着融脂作用,Leptin被释放进血液中,并向大脑发出人体足够进食的信号。然而,大多数超重的人群在他们的血液中有高水平的Leptin..其它分子的提示也对饱食有足够的感觉并对人体体重有影响。
Leptin的发现已经使控制重量的分子研究进入研究的热潮,正在进行的基础研究发现整个网络信号促成激素和其它重要的角色。鼠已被确认为研究人类肥胖的非常有用的模型,并有助于开始阐明帮助控制人体重量的成分。由于有效减肥的市场是巨大的,药品公司正在和基础研究科学家并肩工作以便在控制人体重量的困惑中找到可能的药物靶点。

第九节 血红蛋白尿症
夜间发作的血红蛋白尿(PNH),其独特性迷惑血液家已一个多世纪。PNH是以减少大量红细胞并出现血尿、血浆为特证的,在睡觉后明显。PNH主要与血栓的高危险危害有关,最普遍的血栓出现在腹腔内的大静脉中。大多病人死于血栓。
在PNH血细胞中缺少一种称之为PIG-A的酶,它需要细胞抗基的生物合成。一部分在细胞外面的蛋白质经常被糖基磷基醯肌醇(GP1)抗基束缚于至细胞膜上。如果PIG-A缺乏的话,保护细胞免受血液中结构成分破坏的表面蛋白不会被固定,因此就缺少束缚能力,所以血细胞就被分解了。
在X-染色体上发现PIG-A基因。PNH虽然不是遗传疾病,但是一种遗传障碍,一种获得性遗传障碍。受影响的血细胞克隆通过改变PIG-A至全面降低红细胞、白细胞(包括淋巴细胞)和血小板。血中不正常红细胞的比例决定了疾病的危险程度。

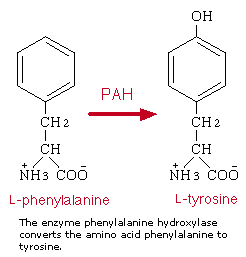
第十节 苯基酮尿酸症
苯基酮尿酸症是一种由缺乏苯基丙氨酸酶所引起的代谢遗传疾病。酶的缺乏导致精神发育迟缓、器官损坏和姿态异常。母亲患PUK严重地危及妊娠。
传统的PKU是一种常规染色体隐性遗传疾病,它是由在第12条染色体上发现的苯基丙氨酸(PAH)两个等位基因突变引起的。在男孩中,PHA把苯基丙氨酸转化为酪氨酸。PHA基因的两对突变意味着酶失去活性或失去效率,使孩子体内苯基氨基酸浓度达到毒素水平,在某些情况,PAH的突变将产生一种称之为高苯基丙氨酸的表现型PKU温和形式。两种疾病是在PHA位点上不同突变的结果;在这种情况下,病人是PAH两种突变的杂合(基因的每一个拷贝有不同的突变),温和突变将占优势。
PKU的形式曾在鼠中发现,这些模型组织正在帮助更好掌握这种疾病,并找到医治这种疾病的方法。仔细的膳食调理会使儿童获得正常生活,有这种病的母亲能生产出健康的孩子。
第十一节 雷弗素姆病
雷弗素姆病是一种罕见的隐性遗传的脂质代谢病,临床表现为慢性神经性炎、肌肉协调萎缩、色素性视网膜炎(渐进性视觉疾病)和共济失调。雷弗素姆病是以植烷酸在原生质和组织中的累积为特点的。植烷酸是叶绿素成分植醇的衍生物。
1997年,确认了雷弗素姆病的基因,并定位第十条染色体上,基因的蛋白质产物PAHX是一种为植烷酸代谢所需要的酶,雷弗素姆病已经损坏PAHX-植烷酸水解酶,由于人类PAHX包含靶向过氧化酶的PTS2定位序列,所以可认为雷弗素姆病是一种过氧化酶障碍。
我们的身体不能合成植烷酸,我们必须从我们所有的食品中获得它,因此,加强用膳食来治疗植烷酸的缺损能获得有效的果。

第十二节 丹吉尔病
丹吉尔病(TD)是一种胆固醇转化的遗传障碍疾病,它由弗吉尼亚海岸丹吉尔孤岛而得名。TD首次是在岛上一个五岁儿童身上确认,他有橙色扁桃体的特点,非常低的高密度脂蛋白(HDL)或富胆固醇和肝脾肿大。
TD是由9q31染色体上ABC1(束缚ATP的盒)的基因突变所致,ABC1编码一种帮助细胞摆脱过量胆固醇的蛋白质,由血中的HDL微粒接纳过量的胆固醇并把它携带至肝脏,经肝脏加工胆固醇被整个身体细胞备用,患有TD病的个体不能从细胞中消除胆固醇,导致在扁桃体和起它器官中类积。这一重要的胆固醇输送基因的发现可以导致掌握HDL水平及在美国与主要危险冠状动脉病之间的相反关系。开发调节HDL水平的新药不仅帮助患TD的病人而且能帮助更多有HDL缺陷家属史的病人。

第十三节 泰—萨病
一种和Ashkenazi Jews有关的遗传性代谢障碍泰-萨病也已在Ouebec东南的法国加拿大人,Louisiana西南的Cajuns人和世界的其他人群中发现。泰-萨病表现的严重性从婴儿到青少年随年令不同有所变化,形成麻痹、痴呆、视觉缺损、和早期死亡,或在漫长的成年期形成神经功能性障碍和精神病。
泰萨病是一种由第15条染色体上等位基因(HEXA)HEXA突变引起的常规染色体隐性疾病。HEXA是Beta-己糖胺酶A的alpha-亚基,它发现在通过细胞为再循环降解大分子的溶酶细胞器中。正常情况下,beta-己糖胺A帮助去降解称之为GM2神经节苷脂的脂质,但是,泰-萨病患者缺乏或只有少量的这种酶,就造成GMZ神经节苷脂在神经中的过量堆积,从泰-萨病表现的各种不同病情中看到神经变性进展首先取决于GM2神经节苷脂累积的速度和程度,其次取决于存在于人体中功能性beta-己糖胺酶的水平。
虽然由于泰-萨病鼠因为分解GM2神经节苷脂具有较少的变化途径而限制了它的用途,但仍然为泰萨病开发了鼠模型。随后对泰-萨病进行的一种神经节苷脂合成抑制剂治疗充满希望。由于对神经节苷脂危险性在婴儿出身前不清楚,因此对患病婴儿采用这种或那种方法治疗的有效性是非常有限的。逆转这危险的困难将使得很难为患病婴儿开发一种有效的治疗方法。然而,后一种治疗泰萨病的措施是有希望的,把这样的治疗方法与DNA和酶的荧光篩选程序在使用中结合起来将导致最终控制这种疾病。
