http://www.100md.com
中国医学论坛报
 |
病历摘要
患儿,女,9岁,因反复发热、咳嗽、面色苍白5个月,口腔溃疡3个月入院。患儿于入院前5个月无诱因出现发热,体温39℃,伴阵发性咳嗽、咳痰、面色苍白、巩膜及皮肤黄染,于当地医院住院治疗。查体双肺呼吸音粗,未闻及干湿罗音,肝脏肋下4.5 cm,脾脏肋下3 cm。血红蛋白49 g/L,网织红细胞12%,Coomb试验阳性,X线胸片示右中叶肺炎和胸膜炎,诊断为“支气管肺炎合并胸膜炎、自身免疫性溶血性贫血”。予丙种球蛋白、氢化考的松、第三代头孢菌素治疗后,患儿体温降至正常,血色素有所上升(具体不详),遂将氢化考的松改为泼尼松口服15天后出院。出院时患儿X线胸片示胸腔积液消失,肺炎无变化,患儿仍咳嗽、咳痰。于入院前3个月,患儿咳嗽、咳痰加重,面色苍白、巩膜及皮肤黄染再现,且合并顽固性的口腔溃疡灶,于同一医院诊断“支气管肺炎、免疫性血管炎”并住院治疗,予甲泼尼龙静点3天,患儿血色素由54 g/L升至108 g/L,后改用泼尼松口服16天好转出院,出院时体温已降至正常。但出院后患儿口腔溃疡面积逐渐增大,咳嗽、咳痰无明显好转。于入院前10天,患儿再次发热,体温38.4℃,为进一步诊治收入我院。
既往史 患儿易患感冒、支气管炎、肺炎。2年前曾患“脑囊虫”和“细菌性痢疾”。
入院查体 体重21 kg,P 120次/分,R 24次/分。发育正常,营养中等,全身皮肤未见黄染、淤点、淤斑,浅表淋巴结未及。口唇无苍白,左侧颊黏膜肿胀、溃烂、有白色脓苔覆盖,无触痛,咽红,扁桃体Ⅰ度肿大。双肺呼吸音粗,右肺呼吸音稍低,可闻及痰鸣音。心音有力,律齐,未闻及杂音。腹平软,未及包块,无压痛及反跳痛,肝脾未触及。神经系统查体未见异常。
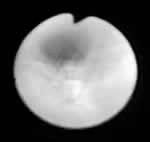
实验室检查 血常规:WBC 4.3×109/L,N 59.1%,L 18.5%,Hb 121g/L,PLT 333×109/L。CRP、ESR均正常。自身抗体阴性,T细胞亚类 CD3 41%(正常66%~76%) CD4 25%(34%~47%) CD8 16%(22%~31%) CD4/CD8比例正常。Ig系列:IgA、IgG、IgE正常,IgM 0.18 g/L正常0.63~2.77 g/L。外院3次Ig系列均提示IgA、IgG 、IgE正常,IgM 均低下,分别为0.16 g/L、0.08 g/L和0.12 g/L。胸部X线显示:双肺纹理多,两下肺中内带片状及网状阴影,右下肺为著。印象:两下肺实质和间质浸润,伴少许通气不良。胸部CT:左上叶和右中叶实变,右中下叶支气管扩张。纤维支气管镜显示:广泛支气管内膜慢性炎症,左上叶和右中叶为著,呈鱼骨刺样改变(见图)。
病例讨论
郭琰医师(内科) 本病例特点:19岁学龄女童,病情反复,病史5个月。(2)主要表现为反复发热、咳嗽、溶血性贫血、口腔溃疡。(3)既往易患呼吸道感染,1998年曾患“脑囊虫、支气管肺炎、细菌性痢疾”。(4)查体左侧颊黏膜肿胀、溃烂,有白色脓苔覆盖,无触痛。双肺呼吸音粗,右肺呼吸音稍低,可闻及痰鸣音。(5)辅助检查,血色素正常,自身抗体阴性,T细胞亚类 CD3 CD4 CD8均减低,血清IgM 低于正常。胸片示两下肺实质、间质浸润,伴少许通气不良。本患儿此次入院虽以呼吸道感染症状为主,但结合其既往曾有多次细菌、病毒、寄生虫感染,且体液及细胞免疫功能均不正常,考虑为免疫缺陷病及自身免疫性疾病。
曾津津医师(放射科) 患儿于入院前5个月X线胸片示两肺肺炎(右中为主)合并胸膜炎、胸腔积液,心影增大。前3个月X线胸片仍有右中叶肺炎,入院前15天X线胸片示双肺纹理粗多,胸腔积液吸收,心影大小正常,右中叶肺炎未吸收。胸部CT示双肺广泛纹理粗多,两下肺和右中叶小囊状改变,支气管壁增厚,走向紊乱,右中叶有致密楔形阴影,纵隔内未见肿大淋巴结。根据X线胸片改变考虑双肺支气管扩张合并右肺中叶不张。
赵顺英医师(内科) 该患儿的诊断考虑(1)支气管扩张 据患儿反复发热、咳嗽、咳脓痰,痰量较多,胸片提示右中叶反复或迁延肺炎,胸部CT示两下肺支气管囊状改变,伴右中叶不张,纤维支气管镜见支气管内大量脓性分泌物,呈鱼骨刺样改变,故支气管扩张诊断成立。(2)口腔黏膜感染 据患儿左颊黏膜肿胀、溃烂,有白色脓性分泌物覆盖,故口腔黏膜感染诊断成立。(3)选择性IgM缺陷病 患儿支气管扩张与反复下呼吸道感染有关,此外病史中有两次自身免疫性溶血,考虑有体液免疫功能缺陷,结合4次(外院3次,本院1次)实验室检验均提示血清IgM<20 mg/dk,其他类别的免疫球蛋白水平均正常,故诊断选择性IgM缺陷病。鉴别诊断要除外系统性红斑狼疮,因患儿存在口腔溃疡、自身免疫性溶血和肺部病变,并且选择性IgM缺陷病可合并系统性红斑狼疮,故应考虑患儿也有系统性红斑狼疮,但2次自身抗体阴性,也无其他表现,未达到系统性红斑狼疮的诊断标准,故可除外。治疗上应输注免疫球蛋白补充IgM,同时予抗生素对症治疗。选择性IgM缺陷病有家族史,应请其父母检查Ig系列。
申昆玲医师(内科) 选择性IgM缺陷病可能是一组病因不同的综合征,其发病机制还不清楚,但无论T细胞或B细胞的缺陷均可致IgM合成障碍,故T细胞在部分病人正常,而另一部分病人不正常。本病可有各种临床表现,包括反复呼吸道、泌尿道感染,播散性传染性软疣、慢性湿疹、异位性皮炎、脂溢性皮炎、自身免疫性贫血和系统性红斑狼疮等。本患儿有反复呼吸道感染、自身免疫性贫血,口腔溃疡迁延不愈,多次检验血清IgM< 20 mg/mk其他免疫球蛋白的水平正常,故可以诊断选择性IgM缺陷病。
江载芳教授(内科) IgM是胎儿最早合成的一种免疫球蛋白,不能通过胎盘由母体输入。在正常情况下,胎龄10.5周左右开始自身合成,出生时含量约为成人的10%,此后合成速度骤增,1~2岁左右达到成人水平。选择性IgM缺陷病是一种原发性免疫缺陷病,定义为血清IgM低于20 mg/dk,而其他免疫球蛋白类别水平正常,T细胞可正常或不正常。选择性IgM缺陷病发病机制尚不明,其发病率为0.03%~1.0%,表现为反复或严重的感染或可无任何症状。本例为国内首例,国外仅报道3例。本病例诊断延迟的原因可能与本病发病率低,临床医师对其认识不够有关。因此即使检查发现异常,也未曾考虑此诊断,仍以“二元论” 诊断该病。
这一病例诊断给我们的启示为,对于反复或迁延呼吸道感染的儿童(可合并其他系统器官感染)一定要考虑基础疾病如原发性免疫功能缺陷、纤毛不动综合征、呼吸道先天畸形等。体液免疫功能缺陷患儿易合并自身免疫性疾病和肿瘤性疾病,结合本例患儿反复感染及合并自身免疫性贫血和顽固性口腔溃疡,考虑其有体液免疫功能缺陷,结合检验结果,最后明确诊断为选择性IgM缺陷病。
目前有学者认为儿科的各系统感染性疾病、自身免疫性疾病、过敏性疾病和各类型的肿瘤等无不与免疫功能紊乱息息相关。通过这个病例的诊断,提示我们需要注意免疫缺陷病。反复或严重的感染常是怀疑本病的线索,实验室检查结果对于诊断和分类都至关重要。