脑白质营养不良
http://www.100md.com
中华儿科网
 |
 |
一、概述
脑白质营养不良在儿科学领域系一相对较陌生的临床问题,多数儿科医师对这一问题缺乏系统了解。笔者也常常被问及对此类疾患的诊断、治疗问题。带着这些问题和自己临床工作中的诸多疑问,作者查阅了相关文献,特别是我国著名小儿神经病学家左启华教授、吴希如教授新近的著作,结合自己肤浅的临床体会,不揣冒昧,录下拙文,供同道们参考。不当之处,还望大家批评。
脑白质营养不良(leukodystrophy)是指遗传因素所致的中枢神经系统正常髓鞘生长受累的疾病。包括多种遗传病所引起的脑白质髓鞘异常,例如:溶酶体病(异染性脑白质营养不良-MLD等),过氧化物体病(肾上腺脑白质营养不良-ALD等),线粒体病(脑神经肌胃肠病-MNGIE等),髓鞘蛋白编码基因缺陷(Pelizaeus-Merzbacher病),氨基酸、有机酸病(PKU、丙酸血症等),以及其他不明原因的脑白质病(Alexander病等)。随着MRI技术在小儿神经临床的应用越来越多,脑白质病变的诊断明显增多。其中有些属于已知遗传或生化机制的脑白质病,属脑白质营养不良范畴;有些则属于未知病因的脑白质病,其中部分是遗传病,也属于脑白质营养不良,其遗传及生化基础尚待探讨;而另一些则由于免疫、炎症、环境等非遗传性获得性因素所致,属于脑白质脱髓鞘病变。
, 百拇医药
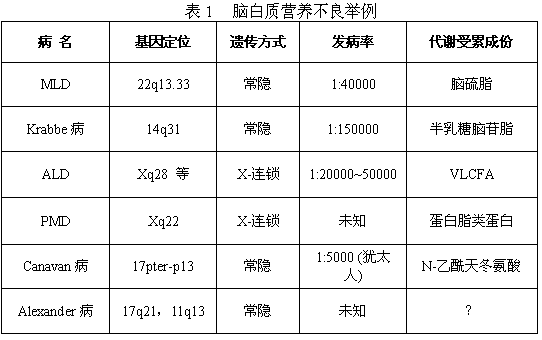
近年由于医学分子生物的快速进展,一些既往较少认识的脑白质营养不良的基因已经定位克隆,其基因产物的性质已经明确。对这些疾病的临床生化诊断、产前诊断,携带者检出及遗传咨询等,在不少发达国家中均已可进行。临床相对常见的部分脑白质营养不良见表1。
神经病理研究证实该组疾病多数属于髓鞘形成障碍疾病(dysmyelinating disease)但其中肾上腺脑白质营养不良兼有脱髓鞘病(demyelinating disease)特点:Canavan病则为髓鞘破坏(myelinolytic)疾病,海棉样变不只累及白质,也累及灰质。
脑白质营养不良的临床表现以逐步进展为特点,早期症状往往易被忽视。原本正常的婴儿或儿童,可逐渐发生肌张力、姿势、运动、步态、语言、进食动作、视觉、记忆学习、行为思考能力等方面的改变。这些征候可逐渐加重,病情进展速度在小儿时期发病者较快。
诊断主要依据:1.临床特点;2.阳性家族史;3.特异性生化检查;4.神经影像学检查。一些发达国家对小儿脑白质营养不良的临床生化与病理形态学诊断途径分几个层次进行:1.一线生化检查有CSF、皮质醇 ACTH实验;2.一线形态学检查有外周淋巴细胞(或脑活体组织)中沉积物;3.二线生化检查有血浆中极长链脂肪酸(VLCFA),尿中硫脂,白细胞溶酶等;4.二线形态检查有皮肤、神经、肌肉或脑活体标本等;5.三线生化检查为分子遗传学方法。
, 百拇医药
二、常见脑白质营养不良的诊断与治疗
(一)异染性脑白质营养不良(metachromatic leukodystrophy,MLD)
MLD又称为脑硫脂沉积病(sulfatidosis),常染色体隐性遗传,是芳基硫酸脂酶A缺陷所致的髓鞘形成不良。由于编码溶酶体芳基硫酸脂酶A(arylsulfatase A,ASA)的基因MLD突变所引致,MLD位于22q13.33,其突变种类较多;大致可分为两组:I型突变的患者不能产生具有活力的ASA,其培养细胞中无ASA活性可测得;A型突变患者则可合成少量具有活力的ASA。患者的表型取决于其基因突变的种类:I型突变的纯合子或具2个不同I型突变者在临床上表现为晚期婴儿型;具有I型和A型突变各一者为青、少年型;而2个突变均为A型时,则呈现为成年型。少数本病患者,特别是青少年型的发病不是由于MLD突变所致,其ASA活力正常,这是由于患者缺少一种溶酶体蛋白,硫酸脑苷酯激活因子(SAP1)所造成的。这类患者亦称为"激活因子缺乏性异染性脑白质营养不良"。
, http://www.100md.com
按起病年龄及临床征象, MLD可分为晚婴型、幼年型和成年型3型。
晚婴型最多见,占全部病例的60%~70%,其发病率约为1/4万,初生时正常,85%发病前已能正常行走。多在2岁左右起病。早期步态异常,共济失调,斜视,肌张力低下,自主运动减少,腱反射引不出,神经传导速度减慢。后者是由于末梢神经受累之故。中期智力减退、反应减少、语言消失、病理反射阳性、不注视、瞳孔对光反应迟钝、可有视神经萎缩。晚期呈去大脑强直体位,偶有抽搐发作。有球麻痹征。病程持续进展,多在4~8岁间死于间发感染。
晚发型(青少年型和成人型)发病年龄自3~10岁至青春期、甚至成人期不等,临床表现不一。起病时也以进行性行走困难为主,伴有腱反射减退、神经传导速度降低等外周神经受累表现;发病年龄较晚的青少年或成年人常先有学习或工作成绩下降、行为异常、认知障碍等,然后才出现共济失调等动作异常和锥体束征。本型病程约为5~10年。
, 百拇医药 本病的确诊依据是ASA活力检测,但在少数有典型症状而ASA活力正常情况时,则应考虑激活因子缺乏性异染性脑白质营养不良的可能性。
本病患者在症状尚未出现以前可考虑进行骨髓移植,以延缓或终止病情发展;对神经系统已有广泛病变者尚无满意治疗方法。
(二)肾上腺脑白质营养不良
肾上腺脑白质营养不良在遗传方式上可分两种类型。一种是较多见的X连锁遗传(X-linked adrenoleukodystrophy,XLALD或简称ALD);另一种是常染色体隐性遗传,发生于新生儿,称为新生儿肾上腺脑白质营养不良(neonatal adrenoleukodystrophy,NALD)。
肾上腺脑白质营养不良的诊断依靠以下检查:①CT和MRI;②电生理检查,儿童ALD早期诱发电位和神经传导速度正常。成人AMN时神经传导速度减慢,脑干听觉诱发电位有异常;③脑脊液,ALD大多正常,可有蛋白和细胞数稍增高。NALD常见脑脊液蛋白增高;④血浆和皮肤成纤维细胞中VLCFA增高,特别是C26脂肪酸增高,C26/C22比值增加,有诊断意义;⑤在发生肾上腺皮质功能不全的阿狄森氏危象时,血中皮质醇减低,在不发生危象时, ACTH刺激试验也能发现肾上腺代偿储备减少。对于男性Addison病,即使未见神经系统症状,也应检测VLCFA,以免漏诊。
, 百拇医药
1.ALD 病理特点是中枢神经进行性脱髓鞘以及/或肾上腺皮质萎缩或发育不良;生化代谢特点是血浆中极长链脂肪酸异常增高;细胞中过氧化物酶体有结构的或酶活性缺陷,故属于过氧化物酶体病(peroxisomal diseases)。ALD临床分为六个类型(表2):1)儿童脑型;2)青春期脑型;3)成人脑型;4)肾上腺脊髓神经病型;5)Addison病型;6)无症状型。杂合子女性也可出现症状,约20%~30%可以发展成类似AMN综合征,但病情较轻,发病较晚,很少见肾上腺质皮质功能不全。在我科报告的29例中,22例儿童脑型、4例青春期脑型、1例肾上腺脊髓神经病型、1例Addison病型和1例无症状型。
激素替代治疗对ALD患者肾上腺素皮质功能不全有效,但不能改善神经系统症状。饮食治疗结合服用Lorenzo油,能使血浆中的C26:O水平降为正常。尽管生化改变令人鼓舞,但临床效果却不理想。骨髓或脐血干细胞移植主要适应于影像学异常明显而神经症候轻度的脑型患儿,可以重建酶活性,改善临床症状,能持久提高认知功能,改善脑磁共振和波谱分析异常程度。但骨髓移植本身有一定的病死率,且价格昂贵,供体困难,随着骨髓移植技术的提高和无症状ALD的早期检出,骨髓移植可望有很好的治疗前景。对症治疗也很重要,包括功能锻炼、调节肌张力和支持延髓功能,鼻饲喂养加强营养,止惊等。
, http://www.100md.com
2.NALD 病理改变严重,脑白质广泛脱髓鞘,灰质亦有轻度变性。可见含脂类的巨噬细胞浸润。肾上腺皮质萎缩,胞浆内有板层状包涵体。患儿肝细胞过氧化物酶体的数目和体积减少。肝大,胆道发育不良。新生儿期首发症状为肌张力减低,惊厥,发育迟缓。可有内疵赘皮、颜面中部发育不良、上睑下垂等。可有肝大。常见白内障碍、眼震、色素性视网膜病。多数病儿在1岁内可有一定程度的发育进步,但以后发育倒退,进行性痉挛性瘫痪,震颤,共济失调,听觉和视觉障碍。有的可见肾上腺皮质功能不全的症状。多在5岁以内死亡。脑脊液常见蛋白增高。诊断靠生化检查。血浆和成纤维细胞的VLCFA水平增高,血中植烷酸增高,六氢吡啶羧酸增多,缩醛磷酸(plasmalogen)减少。在临床上应与脑肝肾综合征(Zellweger病)相鉴别。后者也是常染色体隐性遗传的过氧化物酶体病,但病情更严重,颅面畸形明显,神经系统发育不良,有肝硬化,多发性微小肾囊肿,多在一岁以内死亡。
(三)球形细胞脑白质营养不良(globoid cell leukodystrophy)
, 百拇医药
球形细胞脑白质营养不良(globoid cell leukodystrophy)又名Krabbe氏病,是常染色体隐性遗传病,致病基因位于14q31。其基本代谢缺陷是半乳糖脑苷脂-β-半乳糖苷酶的缺乏,致使半乳糖脑苷脂蓄积于脑内。半乳糖脑苷脂是髓鞘的重要成分,由于酶的缺乏而髓鞘不能代谢更新,因而神经系统有广泛的脱髓鞘,脑白质出现大量含有沉积物的球形细胞。
本病的婴儿型较多见,3~6个月起病,开始有肌张力减低,易激惹,发育迟缓,对声、光、触等刺激敏感。以后肌张力增高,腱反射亢进,有病理反射。末梢神经受累时,则腱反射减低或消失。智力很快减退,常有癫痫发作。视神经萎缩、眼震、不规则发热也是本病特点。有时有脑积水。肝、脾不大。病程进展较快,最后呈去大脑强直状态,对外界反应完全消失,常在2岁以内因感染或球麻痹而死亡。晚发型多在2~5岁起病,主要表现为偏瘫、共济失调、视神经萎缩,以后出现痴呆、癫痫发作。多在3~8岁间死亡。
实验室检查可见脑脊液蛋白增高。电泳可见白蛋白和α2-球蛋白增高,β1-和γ-球蛋白减低。晚发型脑脊液多为正常或仅见轻度蛋白增多。神经影像学检查可见脑的对称性白质病变,晚期可见脑萎缩,脑室扩大。末梢神经传导速度在婴儿型均有明显延缓,在晚发型改变不明显。
, 百拇医药
本病确诊依据白细胞或皮肤成纤维细胞的酶活性测定。杂合子的酶活性在正常与患者之间。可进行产前诊断。
本病治疗无特异方法,主要是支持疗法和对症处理。溶酶体酶替代疗法和骨髓移植疗效尚未得到广泛认可,但已有成功病例。
(四)其他
1.Peizaeus-Merzbacher病(PMD) 是X连锁遗传的进行性髓鞘生成不良,可能与(含)蛋白脂类蛋白(proteolipid protein,PLP)的代谢异常有关,致病基因位于Xq22。病理改变主要是脑白质广泛髓鞘缺乏。以前将本病列入嗜苏丹脑白质营养不良范畴,现认为本病时脑白质很少有嗜苏丹物质。婴儿期起病,生后不久可有非节律的、飘动不定的眼震,发育落后。病程约数年至数十年,逐渐进展。可有小脑性共济失调,视神经萎缩,智力落后,不自主运动,痉挛性瘫,癫痫发作。脑脊液正常。本病亦有其他类型,有的在出生时即发病,很快恶化、死亡;有的为中间类型。诊断根据临床特点及家族史。无有效的治疗方法。
, http://www.100md.com
2.Canavan病 可能是常染色体隐性遗传,病理改变主要见于脑白质,充满含有液体的囊性空隙,似海绵状,故也称中枢神经海绵样变性。未见髓鞘的分解产物,故本病不是原发性脑白质营养不良。脑白质二己糖神经酰胺增多,末梢神经有轴突变性。血浆和尿中N-乙酰天冬氨酸增多。成纤维细胞有天冬氨酸酰基转移酶(aspartoacylase)缺乏,推测脑内也有该酶缺乏,故认为本病与以前报道的N-天冬氨酸尿症(N-aspartic aciduria)可能是同一病种。患儿初生时正常,生后2~4个月开始出现智力发育迟缓,肌张力低下,视神经萎缩。生后6个月开始有明显的进行性头围增大。以后出现癫痫发作,进行性肌张力增高,对声、光、触觉刺激可出现角弓反张。可有舞蹈手足徐动。脑脊液正常。多在5岁以内死亡。有些严重病例在初生时即有肌弛缓,吸吮和吞咽困难,于数周内死亡。也有的起病晚,在5岁以后,表现为进行性痴呆,视神经萎缩,小脑征,锥体束征。诊断根据进行性神经功能衰退,巨头,视神经萎缩,癫痫发作,可考虑本病。CT和MRI可见脑白质有囊样改变。生化检查可见尿中N-乙酰天冬氨酸增多。本病无有效治疗方法。
3.Alexander病 病因尚不明,无特效治疗。婴儿期起病,巨头,智力倒退,痉挛性瘫,癫痫发作。有的病例在儿童期或成年起病。CT检查可见白质弥漫性低密度,额部为著。MRI检查见额部为主的长T1、长T2异常信号,双侧病变弥漫,基本对称。
此外,线粒体病、氨基酸病、有机酸病等遗传代谢病均可伴有脑白质营养不良的病理-临床特点,一般同时具有相应疾病的显著特征。, 百拇医药