关键词:聋;体层摄影术,X线计算机
【摘要】 目的 探讨先天性感音聋的CT诊断价值。 材料与方法 搜集59例先天性感音聋患者,均采用颞骨轴位高分辨CT扫描。 结果 根据其影像学表现将其分为5型:Ⅰ.Michel型,8耳;Ⅱ.Mondini型,15耳;Ⅲ.前庭导水管扩大畸形型,33耳;Ⅳ.内耳道发育畸形型,2耳;Ⅴ.Alexander型和Seheibe型,54耳。值得指出的是多数先天性内耳畸形可伴有或单独存在前庭导水管扩大畸形。 结论 CT对诊断先天性感音聋有重要价值。
CT Diagnosis ofCongenital Sensorineural Hearing Loss
LI Yihong, WEI Jingguo,JIANG Yuexing
(Department of Radiology, Henan Provincial Crops Hospital, Chinese People's Armed PoliceForces, Zhengzhou, Henan Province 450052, P.R.China)
【Abstract】 Objective Toevaluate CT in diagnosing congenital sensorineural hearing loss (CSHL). Materials andMethods Axial high resolution CT scanning of the temperal bone was performed in 59 caseswith CSHL. Results According to their imaging findings, CSHL could be divided intofollowing five types: type Ⅰ, Michel malformation (n=8); type Ⅱ, Mondini malformation(n=15); type Ⅲ, enlargement of vestibular aqueduct (n=33); type Ⅳ, developmentaldeformity of internal acoustic meatus (n=2); type Ⅴ, Alexander malformation and Scheibemalformation (n=54). In most cases, the enlargement of vestibular aqueduct was the onlymalformation or an accompanied malformation. Conclusion CT plays an important role indiagnosing CSHL.
【Key words】 Deafness Tomography,X-ray computed
近十年来,随着CT技术的提高,颞骨薄层扫描及特殊骨重建算法的应用,能显示更小的骨性解剖结构而不会有任何的重叠,弥补了常规X线或体层摄影的不足,从而为颞骨先天畸形提供了有价值的形态学资料,而且90%以上的病例横轴位切面像即能提供所需的全部信息[1] 。笔者搜集本院和外院1994年7月~1998年9月连续患者59例共112耳先天性感音聋的CT所见,并结合文献,探讨本病的CT诊断问题。
1 材料与方法
搜集先天性感音聋且临床电测听或脑干听觉诱发电位反应为中度以上感觉神经聋者59例共112耳,男32例,女27例。年龄10~24岁,其中9岁以下51例。家系调查,4例有阳性家族史,合并先天性外耳道闭锁2例,狭窄1例。判定方法:正常与异常判定测量按宫娟等[2] 报道的方法进行。
使用西门子Somato CR机对所有病例仅行颞骨横断薄层扫描,并用骨重建算法。所有患者均取仰卧位,摆位力求两侧对称,对不能合作者,扫描前口服10%水合氯醛3~10ml,患儿入睡后扫描。以听眶上线为基线,层厚2mm,间隔1~2mm。所有图像均经2位高年资医师盲法读片,独立作出诊断,以2个相同或相近的诊断为该病的常规诊断。
2 结果
59例中31例56耳有内耳畸形,其中合并外耳道闭锁1耳,听骨链畸形3耳,鼓室不含气3耳,内耳道缺如3耳、狭窄3耳、扩大2耳,前庭导水管扩大9耳,耳蜗导水管扩大1耳;单纯前庭导水管扩大33耳;1例只有两内听道扩大;1例仅有双中耳炎伴胆脂瘤形成;余26例52耳CT无异常所见(附表)。
附表 先天性感音聋CT表现
| 畸形类型 | 伴其他异常 | 例数 | 耳数 |
| 内耳完全未发育,岩骨小,耳蜗、前庭、半规管均未发育或发育差 | 1例1耳外耳道闭锁,2例2耳听骨链畸形,1例2耳鼓室气化不良,1例1耳内耳道狭窄,2例3耳内耳道缺如,1例2耳乳突气化不良 | 5 | 8 |
| MondiniⅠ型:耳蜗未发育,前庭、半规管发育不良 | 1例1耳鼓室气化不良,1例1耳听骨链畸形,1例1耳前庭导水管扩大,1例1耳内耳道狭窄 | 3 | 5 |
| MondiniⅡ型:耳蜗发育不良,可累及前庭、半规管 | 2例3耳前庭导水管扩大 | 2 | 3 |
| MondiniⅢ型:耳蜗正常,前庭扩大,半规管发育不良 | 3例5耳前庭导水管扩大,1例1耳内耳道狭窄,1例1耳耳蜗导水管扩大 | 4 | 7 |
| 前庭导水管扩大 | 17 | 33 | |
| 内耳道扩大 | 1 | 2 | |
| 外、中、内耳及颞骨无异常 | 26 | 52 | |
| 外、内耳无异常,双中耳炎伴胆脂瘤 | 1 | 2 |
3 讨论
先天性感音聋的群体发病率约为1/2000~1/6000[3] 。其病因复杂,有遗传、药物、感染等因素,本组有阳性家庭史4例,母亲孕期服药史5例,病毒感染史9例。由于胚胎原基与中、外耳不同,内耳畸形可单独存在,但也可伴中、外耳畸形,多为常染色体隐性遗传,因此临床诊断易发生漏、误诊。颞骨薄层CT检查对此起到了关键性的诊断作用,它可为不同原因的神经性耳聋提供早期病因诊断依据。现结合文献及本组资料将内耳先天畸形和异常归纳为以下5种类型。
3.1 Michel型
此型是内耳最严重的畸形,它指的是内耳完全未发育,表现为岩骨发育小,耳蜗、前庭及半规管均未发育(图1),有的可呈硬块(图2),有的融合为一小腔或大空腔(图3),同时伴内耳道缺如、狭窄或中、外耳畸形。该型通常为常染色体显性遗传[3] ,为胚胎第25天以前的发育障碍[4] 。本组共8耳(单侧2例,双侧3例),同时伴内耳道缺如3耳,狭窄1耳,鼓室不含气2耳(图1),听骨链畸形2耳,外耳道闭锁1耳,乳突气化不良2耳(图1)。

图1 两岩骨发育小,耳蜗、前庭、半规管未发育,两鼓室不含气(▲),两乳突气化不良

图2 左耳蜗、前庭、半规管未发育,呈一硬块(▲),右耳蜗2回,其前庭、半规管未发育

图3 两耳蜗、前庭、半规管未发育,左侧融合为一小腔,右侧融合为一大腔
3.2 Mondini型
此型分3类。第1类:畸形严重程度仅次于Michel型。特点是耳蜗未发育,前庭发育不良,多表现为前庭扩大,半规管小、变粗,尤以外半规管明显(图4)。此型共5耳(单侧1例,双侧2例),同时伴内耳道狭窄1耳,鼓室不含气、听骨链畸形各1耳,前庭导水管扩大1耳。第2类:Mondini型。此型为内耳发育不全聋,主要影响耳蜗,可累及前庭。耳蜗发育不良表现为耳蜗只有1回(图5)、1.5回、2回(图5),或形态不正常,如小、扁平等,个别耳蜗与前庭分化不良。累及前庭表现为前庭扩大、半规管短小或未发育(图2),以外半规管最常见。该型为常染色体显性遗传,为发生在妊娠6周的发育障碍[3] 。此型共3耳(单侧1例,双侧1例),累及前庭导水管扩大3耳。第3类:前庭型(不典型的Mondini型)。耳蜗骨迷路正常,表现为前庭、半规管的发育不良或畸形共7耳(单侧1例,双侧3例),伴前庭导水管扩大5耳(图6),内耳道狭窄1耳,耳蜗导水管扩大1耳。

图4 左耳蜗未发育,前庭扩大,外半规管短粗(▲),两前庭导水管喇叭状扩大
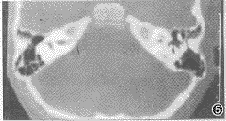
图5 右耳蜗2回,右耳蜗、外半规管发育不良;左前庭、外半规管及内听道未见异常
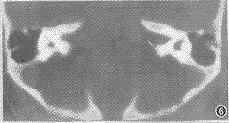
图6 两前庭扩大,外半规管短粗,两前庭导水管喇叭状扩大
3.3 前庭导水管扩大畸形
Vallvassori等[4] 认为前庭导水管扩大是内耳迷路发育完成以后所发生;Vignaud等[5] 认为此种畸形发生于胚胎第90天之后;Tong等[6] 认为该型患者有家族遗传倾向,根据家谱分析这种类型多为常染色体隐性遗传,少部分可能为常染色体显性或多因素遗传,其管口最大径>1.5mm为扩大;国内刘中林等[7] 也有类似观点。本组共33耳(单侧1例,双侧16例),表现为前庭导水管喇叭型扩大(图4、6),其中有些与半规管总脚相通(图7)。

图7 两前庭导水管扩大并与总脚相通
3.4 内耳道发育畸形
包括内耳道缺如、狭窄、扩张。前两者已在前面叙述。本组有1例2耳双侧内耳道扩大,其直径约9mm(图8)。
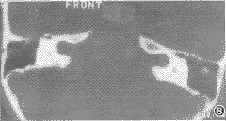
图8 两内听道扩大,直径约9mm
3.5 Alexander和Scheibe型
Alexander型为蜗管发育不全,蜗基底转螺旋器和邻近的螺旋神经节细胞多受影响[3] ,属膜迷路异常。Scheibe型为球囊和蜗管发育不全,此两型均为内耳膜迷路异常,均发生于骨迷路正常形成之后,故骨迷路正常;而膜迷路异常用CT和其他影像手段均不能显示,故CT无异常所见[8] 。本文感音聋组中27例54耳内耳无异常所见(其中1例有双侧中耳炎伴胆脂瘤形成),可能属此两型之一。
总之,颞骨薄层CT诊断先天性感音聋简单、经济、准确,本文将先天性感音聋分为5型,这种细微的分类有利于对先天性内耳畸形进行更深入的研究。此外,CT显示了耳聋患者耳蜗骨迷路正常与否,对了解耳聋与耳蜗畸形的关系,对人工耳蜗植入适应证的选择有帮助。但对耳蜗膜迷路异常不能显示,因此对这类先天性异常疾病的诊断无能为力;另外,横断面CT对上、后半规管不能显示其全貌,所以对上、后半规管发育异常的诊断有限。值得指出的是本文39%(23/59)的先天性内耳畸形伴有或单独存在前庭导水管扩大畸形,可见,前庭导水管扩大较常见,应引起临床医生的高度重视。
参考文献
1,Mancuso AA, Hanafee W. Computedtomography and magnetic resonance imaging of the head and neck. 2nd edition. Williamo& Wilkins, Baltimore, 1985
2,宫娟,兰宝森.先天性感音聋的所见.中华放射学杂志, 1990, 24:154
3,卜行宽,综述.遗传性感音神经性聋.国外医学*耳鼻咽喉科分册,1987, 11:321
4,Vallvassori GE, Clemis JD. The large vestibular aqueduct syndarome. Laryngoscope,1978, 88:723
5,Vignaud J, Ohta F. Tomodensitométrie cranio-encé-phalique. Paris:Vigot,1987, 391
6,Tong KA, Harnsberger HR, Dahlen RT,et al. Large vestibular aqueduct syndrome: Agenetic disease? AJR, 1997, 168:1097
7,刘中林,兰宝森,廉能静,等.前庭导水管扩大的研究.中华放射学杂志,1998, 32:268
8,Mafee MF, Selis JE, Yannias DA,et al. Congenital sensorineural hearing loss.Radiology, 1984, 150:427
(收稿: 1999-08-09 修回:1999-12-22 ) , 百拇医药