 |
关键词:包涵体;肌炎;活检;;病理
【摘要】 目的 探讨包涵体肌炎(IBM)的临床与病理特点。方法 总结3例IBM病人的临床特点,并对肌活检标本进行酶组织化学、组织化学病理和超微病理研究。结果 3例女性病人均在24~36岁发病,其临床特点为以双下肢无力起病,渐累及上肢,远端肢体受累常见。腱反射消失,血清肌酸激酶正常或轻度增高,肌活检光镜检查发现其主要病理改变为镶边空泡纤维,肌浆或肌核内有嗜酸性包涵体,肌内膜炎性细胞浸润和成群萎缩肌纤维。电镜观察发现3例均有肌浆内细丝或管状细丝包涵体,其中1例有核内包涵体。镶边空泡内含淀粉样细丝、髓样结构、絮状无结构物质和其他胞浆分解产物。肌核改变包括异染色质增多、核变大,核内包涵体及核崩解。结论 电镜包埋、半薄切片定位是电镜下寻找包涵体并确诊IBM的关键步骤。肌核改变可能是IBM的病因基础,镶边空泡和肌浆内包涵体有可能来自于崩解的肌核。
A clinical and pathological study of 3 cases of inclusionbody myositis Yan Chuanzhu , Li Danian, Liu Shuping, et al. Department of Neurology, Affiliated Hospital of Shandong MedicalUniversity, Jinan 250012
【Abstract】 Objective The study was toinvestigate the clinical and pathological features of inclusion body myositis(IBM). Method Muscle biopsied specimens from the 3 patients with IBM wereanalyzed with histochemical and enzyme histochemical methods under light microscopy andelectron microscopy. Results The 3 patients whowere all females and aged from 24 to 36 years had an onset with lower limb weakness whichascended to involve the upper limbs. The distal weakness was usually as great as or evengreater than the proximal weakness. Diminished or lost deep reflexes were found in all the3 patients. Light microscopic examination showed that the rimmed vacuolated muscle fibers,the eosinophilic inclusions, atrophic fibers in groups and the inflammatory exudate withinthe endomysia constituted the cardinal pathological characteristics. Under the electronmicroscope, amyloid fibrils or tubulofilament inclusions in the cytoplasms were found inall 3 cases and intranuclear inclusions were found in only one case. Myonuclearalternations consisted of large nuclei, intranuclear inclusions, an excess ofheterochromatin and myonuclear degradation. Conclusion Localization on semisection under the electron microscope might bethe crucial procedure for finding the inclusions and making a definite diagnosis of IBM.Also the results suggested that myonuclear alterations might lead to the basic genesis ofIBM.
【Key words】 Inclusion body Myositis Biopsy Pathology
包涵体肌炎(IBM)是一种不同于多发性肌炎和(或)皮肌炎的慢性炎症性肌病。Yunis等[1] 于1971年首先提出了IBM这一名称,直至1978年,Carpenter等[2] 总结了14例IBM病人的临床病理特点并确立了IBM为一种独立的临床疾病。此后国外有百余例IBM的临床病理报告[3~7] 。国内仅孙畸等[8] 报道1例IBM,但未经电镜证实有包涵体。我们报道3例经电镜证实的IBM,并对其临床、组织病理及超微结构特点进行初步探讨。
资料和方法
一、临床资料
例1 女,24岁,主诉下肢无力3年,进行性加重并上肢无力半年。现不能快走,上楼困难,双小腿变细。无肌痛,无吞咽困难及肌束颤动。体格检查:双侧肱二头肌、岗上肌、岗下肌肌力稍弱,双上肢远端肌力正常,不能仰卧起坐,下肢股四头肌、腓肠肌、胫前肌肌力3~4级,趾伸肌肌力3级。肌张力低,双小腿肌萎缩。四肢腱反射消失。血清磷酸肌酸激酶正常。两次肌电图(EMG)检查分别示神经源性损害和肌源性损害。
例2 女,36岁,主诉腰髋部酸软无力8年,逐渐加重,下蹲后不能站立、上楼困难3年。近1年来出现双手及大腿肌肉跳动。无肌痛及吞咽困难。体格检查:面及颈部诸肌肌力正常,三角肌肌力4级,上肢远端肌力均为3级,双膝、踝关节伸肌肌力3级,屈肌肌力4级,不能仰卧起坐,下蹲后不能自行站起。四肢无明显消瘦,但肌腹消失,肌坚度差。四肢腱反射消失。血清肌酸激酶(CK)为694 U/L。EMG示肌源性损害。
例3 女,25岁,主诉双下肢无力渐累及上肢4年,产后加重2年。现行走无力,上楼困难,全身消瘦,易疲劳,有时小腿僵硬,有酸胀感,无明显肌痛及吞咽困难。查体:头颈部肌肉肌力正常。双上肢远近端肌力4级,下肢股四头肌肌力5级,股二头肌肌力3级,踝关节伸屈肌力均为4级,趾伸屈肌肌力2级,髋腰肌肌力正常。全身肌肉均匀萎缩,行走时腰前倾。四肢腱反射消失。两次检查血清CK值分别为213 U/L和606 U/L。EMG示肌源性损害。
二、方法
一部分活检肌肉经速冻,制成8 μm切片,行苏木素-伊红(HE)、改良高墨瑞(MGT)、还原型辅酶I(NADH-TR)、高碘酸Schiff反应(PAS)、ATP酶(pH10.4、4.65、4.35)、酸性磷酸酶(ACP)和油红“O”染色。另一部分肌肉经3%戊二醛固定,Epon812包埋,制成1 μm切片,甲苯胺蓝染色,光镜下选择含包涵体或空泡的肌纤维进行定位修块,制成超薄切片,双染色,在日本产H-800透射电镜下观察。
结果
一、光镜下病理改变
3例病人肌活检标本光镜下病理改变见表1。其中较具特征性的病理改变是肌纤维内含镶边空泡(rimmedvacuoles)和嗜酸性包涵体。镶边空泡光镜下呈圆形、卵圆形或不规则形态,大小不一。空泡周边常有深染的嗜碱性颗粒聚积。嗜酸性包涵体HE染色下呈均质样红色圆形小体,常位于镶边空泡中央或周边,也可见于肌核内。
表1 3例包涵体肌炎病人的光镜改变
| 例 序 | 包涵体数 | 镶边 空泡 | 成群萎 缩MF | 肌核 改变 | MNC 侵入NF | 肌内膜 MNC | 单个 坏死 | ACP阳 性MF | |
| 浆内 | 核内 | ||||||||
| 1 | 2 | 1 | +++ | ++ | ++ | + | + | - | + |
| 2 | 0 | 0 | + | + | + | - | + | - | - |
| 3 | 1 | 0 | ++ | ++ | ++ | + | + | + | - |
二、超微病理改变
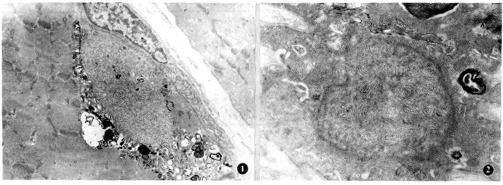
管状细丝包涵体由双股细丝构成,平行排列。细丝包涵体由淀粉样细丝构成,乱杂排列。电镜下见镶边空泡内含大量涡轮状髓样小体、电子致密的黑色小体和一些膜碎片(图1)。肌核改变包括:肌核变大,异染色质增多。有的核膜崩解,只留下一个由电子致密物组成的核的轮廓,核内偶见细丝包涵体(图2)。线粒体改变主要为线粒体嵴排列异常,呈多囊状或不规则排列。3例IBM病人主要超微病理改变见表2。
讨论
IBM是一种慢性炎症性肌病,起病隐匿,进展缓慢。Mendell等[9] 认为IBM的病程至少应超过6个月。本组3例,从起病至确诊,最长者8年,最短者3年。有关IBM的发病年龄,多强调50岁以后好发,但Eisen等[10] 发现,20~30岁和60~70岁为两个发病高峰,分别称为青年组和老年组。Chou[11] 也支持这一观点。本组3例均为20~30岁起病,应属青年组。本组无老年病人可能与病例数少有关。IBM常以四肢近端无力起病,但远端受累也很常见,且有38%的病人远端肌无力程度超过近端[3] 。本组病人均以下肢肌无力起病,远端受累明显,且有选择性累及某些肌肉的倾向。如例3,股四头肌、骼腰肌肌力完全正常,而股二头肌肌力仅Ⅲ级。这与Lotz等[3] 所描述的股四头肌和骼腰肌较易受累并不完全一
表2 3例包涵体肌炎病人的超微结构改变
| 例序 | 包涵体 | 核异常 | 线粒体 异常 | MNC侵 入NF | 毛细血 管异常 | 无结构 物质 | 黑色小体 | 髓样结构 | |||
| 淀粉细丝 | 管状细丝 | 浆内 | 核内 | ||||||||
| 例1 | + | - | + | + | + | + | + | + | + | + | + |
| 例2 | - | + | + | - | + | + | - | - | + | + | + |
| 例3 | + | - | + | - | + | - | + | + | + | + | + |
致,表明受累的肌肉并不固定。腱反射消失是IBM的另一临床特点[3,4,9] ,本组3例病人,腱反射均消失。有学者认为IBM腱反射减退或消失与其经常伴周围神经损害有关[3] ,但本组除1例有1次EMG检查提示神经源性损害外,无其他合并周围神经病的证据。
IBM最具特征性的病理改变是肌纤维内含包涵体。在光镜下观察,有58%的IBM病人可发现嗜酸性包涵体,但每张切片上很少超过3个[3] 。本组例1和例3光镜下发现嗜酸性包涵体。IBM光镜下另一突出的病理改变是镶边空泡纤维。尽管几乎所有IBM均有镶边空泡纤维,但它并非IBM所特有的改变。眼咽型肌营养不良、胫骨前肌营养不良及酸性麦芽糖酶缺乏症也常见镶边空泡,但不伴炎性细胞浸润,可资与IBM区别。IBM在光镜下的其他病理改变,如成群肌纤维萎缩、单核细胞侵入非坏死纤维也常见,其出现率分别为96%和92%[3] 。尽管两者均为非特异性病理改变,但当与镶边空泡同时出现时,强烈提示IBM的可能。
电镜下发现肌纤维内有管状细丝或淀粉样细丝包涵体即可确诊IBM[3,9,12] 。为提高电镜下包涵体的检出率,可先经甲苯胺蓝染色后,选择含空泡的肌纤维,再修块做超薄切片。本组病人开始在未经定位做成的超薄切片上,均未找到包涵体,后经重新做半薄切片定位后,2例在半薄切片光镜下就发现了包涵体,另1例电镜下也找到了包涵体。因此我们认为对IBM的电镜诊断,半薄切片定位至关重要。
肌核的改变被认为可能是IBM原发病变部位[9] ,我们对3例病人的观察发现,IBM一些特征性的病理改变均可能与肌核有关。如:(1)光镜下镶边空泡常位于肌膜下,与肌核的位置相当,且镶边空泡内物质与肌核均为嗜碱性;(2)细丝包涵体既可见于肌核内,也可见于肌浆内,但肌浆内的包涵体几乎总是在镶边空泡内或其周边,包涵体周围多有嗜碱性物聚积;(3)电镜下可观察到处于不同时期崩解的肌核,最早期为异染色质增多,体积增大,继之核膜消失,并崩解成空泡,内含髓样结构或碎片等崩解产物。以上现象提示,镶边空泡和肌浆内包涵体有可能来自于崩解的肌核,后者尚有待于应用细胞化学的方法加以证实。
与多发性肌炎和(或)皮肌炎不同,IBM对类固醇治疗无效[13,14] ,但对静脉用免疫球蛋白治疗可能有效[15] 。IBM一经确诊,不宜再用类固醇治疗,以免出现严重的副作用,使病情恶化。静脉用免疫球蛋白对IBM究竟有无确切疗效尚有待进一步研究予以证实。
作者单位:250012 济南,山东医科大学附属医院神经内科(焉传祝、李大年、刘淑萍、吴金玲、郭斌);烟台市中医医院(张雅萍)
图1 电镜下胞浆内细丝包涵体,周边髓样结构,黑色小体和絮状无结构物质 ×6400
图2 电镜下肌核内细丝包涵体 ×10 000
参考文献
1 Yunis EJ, Samaha FJ. Inclusion body myositis.Lab Invest,1971, 25:240-248.
2 CarpenterS,Karpati G,Helle I,et al. Inclusion body myositis:a distinct variety of idiopathicinflammatory myopathy. Neurology, 1978,28:8-17.
3 LotzBP,Engel AG,Nishino H,et al. Inclusion body myositis.Brain, 1989,112:727-747.
4 SayersME,Chou SM,Calabrese LH.Inclusion body myositis:analysis of 32 cases.JRheumatol,1992,19:1385-1389.
5 MendellJR,Sahenk Z,Gales T, et al. Amyloid filaments ininclusion body myositis.Novel findings provide insight into nature of filaments.ArchNeurol,1991,48:1229-1234.
6 EngelAG,Arahata K.Monoclonal antibody analysis of mononuclear cells in myopathies.Ⅱ:phenotypes of autoinvasive cells in polymyositis and inclusion bodymyositis.Ann Neurol,1984,16:209-215.
7 Ringel SP,Kenny CE,Neville HE,et al.Spectrum of inclusion body myositis. ArchNeurol,1987,44:1154-1157.
8 孙畸,张平.包函体肌炎(附1例报告并文献复习).临床神经病学杂志, 1996,9:240-241.
9 GriggsRC,Askanas V,Dimauro S, et al. Inclusion body myositis and myopathies. AnnNeurol,1995,38:705-713.
10 Eisen A,Berry K,Gibson G.Inclusion bodymyositis:myopathy or neuropathy ? Neurology, 1983,33:1109-1114.
11 Chou SM.Inclusion body myositis:a chronicpersistent mumps myositis? Hum Pathol,1986,17:765-777.
12 Dalakas MC.Polymyositis,dermatomyositis andinclusion body myositis. N Engl J Med, 1991,325:1487-1498.
13 Danon MJ.Reyes MG,Perurena OH,et al. Inclusionbody myositis.A corticosteroid-resistant idiopathic inflammatory myopathy. ArchNeurol,1982,39:760-761.
14 Leff RL,Miller FW,Hicks J,et al.The treatment ofinclusion body myositis: a retrospective review and a randomized,prospective trial toimmunosuppressive therapy. Medicine,1993,72:225-235.
15 Dalakas MC,Dambrosia JM,Sekal EA,et al. Theefficacy of high-dose intravenous immunoglobulin in patients with inclusion body myositis.Neurology,1995,15(Suppl):208.
(收稿:1997-07-13 修回:1998-01-23)
, http://www.100md.com