第九章 弥散性血管内凝血
弥散性血管内凝血(disseminated or diffuse intravascular coagulation, DIC)是指在某些致病因子作用下凝血因子或血小板被激活,大量可溶性促凝物质(soluble thromboplastin)入血,从而引起一个以凝血功能失常为主要特征的病理过程(或病理综合征)。此时微循环中有纤维蛋白性微血栓或血小板团块形成,同时一系列血浆凝血因子被消耗,血小板减少,并有继发性纤维蛋白溶解(纤溶)过程加强。在临床上,DIC患者主要表现为出血、休克、脏器功能障碍和贫血。
由于DIC的发病机制和临床表现比较复杂,因此长期以来,曾经有过许多不同的命名。近年来,某些学者认为,为了更全面地表达此病理过程的变化特点,建议将DIC称为消耗性血栓-出血性疾病(cosumptive thrombohemorrhagic disorders)。本章继续使用弥散性血管内凝血的命名。
第一节 弥散性血管内凝血的原因和发病机制
正常机体的血液呈液体状态,在心、血管内流动不止,同时也不发生血凝。这是由于机体存在着凝血、抗凝血和纤维蛋白溶解系统,它们处于动态平衡状态。其中以凝血过程和纤维蛋白溶解过程最为重要,二者保持着极为密切的关系。
DIC的病因众多,引起DIC的发病机制为复杂,但其中以血管内皮细胞的损伤与组织损伤为最重要。
一、血管内皮细胞损伤、激活凝血因子Ⅻ,启动内源性凝血系统
细菌、病毒、螺旋体、高热、抗原抗体复合物、休克时持续的缺血、缺氧和酸中毒、败血症时的细菌内毒素等,在一定条件下皆可使血管内皮细胞发生损伤,使其下面的胶原暴露。胶原、内毒素等均为表面带负电荷的物质,当无活性的凝血因子Ⅻ与这些物质表面发生接触后,其精氨酸残基上的胍基在负电荷影响下分子构型发生改变,它的活性部分――丝氨酸残基暴露,所以因子Ⅻ被激活(此种激活方式称接触激活或固相激活)。另外,也可能在激肽释放酶、纤溶酶或胰蛋白酶等可溶性蛋白水解酶的作用下,因子Ⅻ或Ⅻa通过酶性水解(酶性激活或液相激活)而生成Ⅻf。胶原等激活因子Ⅻ的过程开始时进行得较为缓慢,但因Ⅻ的碎片(Ⅻf),即激肽释放酶原激活物(predallidrein activator, PKA)可把血浆激肽释放酶原(prekallikrein)激活成激肽释放酶(kallikrein),后者又能反过来使因子Ⅻ进一步活化,从而使内源性凝血系统的反应加速(图9-1)。Ⅻa和Ⅻf还可相继激活纤溶、激肽和补体系统,从而进一步促进DIC发展。

图9-1 血液凝固过程及纤溶系统
以内毒素血症为例,此时,除内毒素可直接激活因子Ⅻ外,内毒素还可引起血管内皮细胞损伤,基底膜暴露后,胶原、胶原与某些糖蛋白的复合物或另一些结缔组织成分也可激活因子Ⅻ。此外,还有某些酸糖脂(acidic glycolipids),硫酸脂(sulfatides),氨基葡聚糖(glycosaminoglycans)或另外一些特殊的因子Ⅻ激活物也可自损伤的内皮细胞释放,因此内源性凝血系统启动。在家兔内毒素引起的DIC中常可有内皮细胞脱落,在循环血中有内皮细胞出现。抗原抗体复合物粘附在肾小球等微血管壁上时,可引起血管内皮细胞损伤。血管炎时也可继发血管内皮细胞损伤,进一步触发DIC。
此外,在内皮细胞受损时,血小板与内皮下结缔组织中的胶原接触后可以产生胶原诱导的促凝活性,此时,因子Ⅺ可不通过Ⅻa而直接被激活,从而推动凝血连锁反应,引起DIC。
表9-1不同的人体组织中凝血因子Ⅲ的含量
| 组织 | 含量 (μ/mg) |
| 肝 | 10 |
| 肌肉 | 20 |
| 脑 | 50 |
| 肺 | 50 |
| 胎盘 | 2,000 |
| 蜕膜 | 2,000 |
二、组织严重破坏使大量组织因子进入血液,启动外源性凝血系统
在外科大手术、严重创伤、产科意外(如胎盘早期剥离、宫内死胎等)、恶性肿瘤或实质性脏器的坏死等情况下均有严重的组织损伤或坏死,所以大量促凝物质入血(表9-1),其中尤以组织凝血活酶(tissue thromboplastin,即凝血因子Ⅲ,或称组织因子)较多。这些促凝物质可通过外源性凝血系统的启动引起凝血。
组织因子是一种脂蛋白复合物,含有大量磷脂。当它进入血浆后。血浆中的钙离子将因子Ⅶ连接于组织因子的磷脂上,形成复合物,后者可使凝血因子X活化为Xa,并与Ca2+、因子V和血小板磷脂相互作用而形成凝血酶原激活物,然后通过与内源性凝血系统后阶段相同的途径,完成凝血的化学反应(图9-1)。
以宫内死胎为例,当胎儿的坏死组织在子宫内滞留超过5周,DIC的发生率可达50%左右,这是因为坏死的胎儿组织释放组织因子,后者大量进入母体循环,启动外源性凝血系统。此外有人证明,当肿瘤组织坏死时,释放出一种蛋白酶,如某些腺癌能分泌一种含有唾液酸的粘蛋白,它可直接激活X因子,从而启动凝血连锁反应。
三、血细胞大量破坏
红细胞大量破坏时常可发生DIC。急性溶血,如大量(>50ml)误型输血、药物引起的免疫性溶血时,抗原-抗体复合物的形成对凝血起主要作用。因为据报道,在蚕豆病中由非免疫因素引起的血管内溶血以及实验性血红蛋白尿等情况下常常不产生DIC。因此,一般认为只有在红细胞大量破坏伴有较强的免疫反应时,DIC才比较容易发生。此外,红细胞大量破坏释出的ADP与DIC的发生有关,因为后者触动了血小板释放反应,使大量血小板第3因子(PF3)入血,促进凝血过程。红细胞膜内大量的磷脂既有直接的促凝作用,又能促进血小板的释放而间接促进凝血过程。
实验研究证明,正常的中性粒细胞和单核细胞内有促凝物质。在内毒素或败血症所引起的DIC时内毒素可使中性粒细胞合成并释放组织因子,同时有大量白细胞在肺血管中停滞,并释放出大量促凝物质(可能就是组织因子),这些物质进入体循环进一步加速了凝血反应,所以肺似乎起了凝血的放大作用。大量促凝物质从崩解的白细胞中释放出来,从肺血管经左心进入主动脉后,肾脏首先受累,因此肾脏微血栓发生率较高,病变程度较重。另外,在病人患急性早幼粒细胞性白血病时,此类白血病细胞浆中含有凝血活酶样物质,当白血病细胞大量坏死或经化疗杀伤时,这些物质就大量释放入血,通过外源性凝血系统的启动而引起DIC。
血小板在DIC的发生发展中起着重要的作用。内毒素、免疫复合物、颗粒物质、凝血酶等都可直接损伤血小板,促进它的聚集。微血管内皮细胞的损伤,内皮下胶原和微纤维的暴露是引起局部血小板粘附、聚集、释放反应的主要原因,这是因为是构成胶原的肽链中,存在着一个与血小板粘附有关的活性部位。血小板表面的糖蛋白Ib(glycoprotein Ib, GPIb)对血小板粘附起重要作用,GPIb通过血浆因子(如Ⅷ相关抗原/von Willebrand因子,Ⅷ/VWF因子)使血小板与内皮下组织粘连。另外,由于血小板膜上的另一些糖蛋白(GPⅡb,GPⅡa)能结合于纤维蛋白原,后者通过与钙离子的连接,在血小板之间“搭桥”,使血小板聚集。血小板发生粘附、释放和聚集后,除有血小板微集物形成(microaggregate formation,图9-2)堵塞微血管外,还能进一步激活血小板的凝血活性,促进DIC的形成。但是在不同病因所引起的DIC中,血小板所发挥的作用并不一致,它可以起原发的作用,如血栓性血小板减少性紫癜,在发病开始时即可由免疫反应等原因使血小板发生聚集,其中PF3(血小板第3因子)能加速凝血酶原的激活,PF4(血小板第4因子)能中和肝素并使可溶性纤维蛋白多聚体沉淀。β-血栓球蛋白也具有促凝作用,从而加速血液凝固,形成微血栓。但是,一般来说,在DIC发病中,血小板多起继发的作用。在外源性凝血系统被激活所致的DIC中,血小板不起主要作用,在内毒素引起的DIC中,血小板对白细胞的促凝机制还有促进作用。实验证明,人类白细胞与内毒素同时孵育后所产的促凝活性可因加入血小板而增强,这可能是血小板膜上的脂蛋白、白细胞及某些凝血因子相互作用造成的。
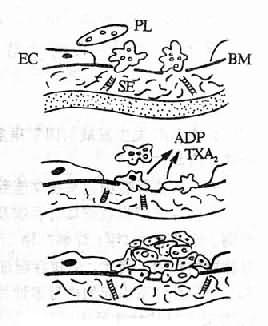
图9-2 血小板微聚物形成机制示意图
PL血小板 EC 内皮细胞
SE 内皮下组织 BM 基底膜
四、其它促凝物质进入血液
一定量的羊水、转移的癌细胞或其它异物颗粒进入血液可以通过表面接触使因子Ⅻ活化,从而激活内源性凝血系统。急性胰腺炎时,蛋白酶进入血液能促使凝血酶原变成凝血酶。毒蛇咬伤时,某些蛇毒如蝰蛇的蛇毒含有一种蛋白酶,它可直接水解凝血酶原形成凝血酶。响尾蛇的蛇毒可直接使纤维蛋白原凝固。抗原抗体反应也可以引起DIC,这可能是抗原抗体复合物能激活因子Ⅻ或损伤血小板引起血小板聚集并释放促凝物质(如血小板因子等)所致。补体的激活在DIC的发生发展中也起着重要的作用。有人发现,给正常动物静脉注射内毒素后,出现动脉血压下降,血小板及纤维蛋白原等凝血因子减少;但如事先耗竭动物的补体,然后再注射内毒素,则该动物血压改变不明显,DIC实验室检查的异常变化轻微,存活率比未去除补体的动物高,由此可见补体系统在内毒素引起的的DIC中也起一定的作用。补体系统激活的产物C3a、C5a可引起组织肥大细胞、血液嗜碱性粒细胞的脱颗粒反应,从而释放5-羟色胺、组胺等物质。组胺能使毛细血管、微静脉等部位的血管内皮细胞收缩,内皮细胞之间的裂隙扩大,内皮下的胶原暴露,促使内源性凝血系统激活。此外,补体系统激活后C3b还可通过人单核细胞上的C3b受体而使凝血因子Ⅲ的释放增多。补体系统还能直接或间接地促进血小板释放PF3。
上述各种所致DIC的机制如图9-3所示。它们常常综合或相继起作用。

图9-3 DIC的发病机制
, http://www.100md.com