消化内科肝肾综合征发病机制研究进展
作者:谭凯 王天才
单位:谭凯 王天才 430030同济医科大学附属同济医院
关键词:
德国医学/990520 肝肾综合征(HRS)是慢性肝病和晚期肝功能衰竭患者的一个常见并发症,它以肾功能衰竭和显著的全身血流动力学异常以及内源性血管活性系统的改变为特征。
晚期肝病患者可发生肾功能衰竭,这在19世纪末就已得到公认。但直到50年代末才出现了详细描述这一综合征的报道。有关HRS理论基础的主要论断是:肾功能衰竭常呈急进性发展并与全身循环的减少有关;尽管肾功能衰竭严重,但这些患者的肾脏没有显著的组织学异常,HRS患者的肾脏移植给慢性肾功能衰竭患者时可使其肾功能恢复正常,而进行肝移植后的HRS患者其肾功能衰竭能逆转[1]。然而,有关HRS的发病机制至今尚未完全阐明,有些观点亦存在较大争议。本文就HRS的病理生理学及其发病机制作一简述。
, 百拇医药
一、病理生理学
显著的肾血管收缩导致肾脏低灌注是HRS的特点。HRS患者体内存在的显著的肾血管收缩已经通过许多检测方法得到证实,包括肾动脉造影术、133氙洗出术、对氨马脲酸清除率及最近应用的多普勒超声检查。应用这些技术已经证明了肝硬化腹水患者肾灌注的变化是连续的,而HRS则是这一系列变化的终点。与在肾血管系统中发现的显著的血管收缩相反,肾外血流动力学以低全身血管阻力和动脉低血压为特征[2]。
(一)血管收缩因子
导致HRS肾血管收缩的潜在介质已经涉及到肾素——血管紧张素——醛固酮系统、交感神经系统和在肾循环中具有血管收缩效应的一些主要系统。这些血管收缩系统的活性在肝硬化腹水病人,尤其在并发HRS的患者是增加的。
1.肾素——血管紧张素——醛固酮系统(RAAS)。Wilkinson等[3]证实,50%~80%的失代偿性肝硬化患者的RAAS活性是增强的,且在HRS患者其活性进一步提高,表明血管紧张素Ⅱ(ATⅡ)在HRS肾血管收缩的发病机制中起作用。有资料表明,用肌丙抗增压素成血管紧张素转化酶抑制剂抑制肝硬化RAAS引起显著的低血压和肾小球滤过率(GFR)的下降,相反,肝硬化患者在灌注ATⅡ后其GFR增加,这可能是ATⅡ导致动脉压增加的结果[4]。因此,认为RAAS对肾功能的影响可能与全身血流动力学变化有关。但另有资料表明,ATⅡ也可产生于肾脏循环内[5],所以肾素—血管紧张素系统亦可能作为局部血管收缩因子参与肝硬化肾血管收缩的发病机制。
, 百拇医药
2.交感神经系统(SNS)。 自80年代以来,人们已经认识到了肝肾神经支配的重要性。研究证实,HRS患者的SNS高度激活[6],其动脉和肾静脉的去甲肾上腺素(NE)浓度与肾血浆流量(RBF)呈负相关[7]。肾脏SNS的激活通过入球小动脉的收缩,导制RBF和GFR的减少及钠潴留。相反,麻醉阻滞腰SNS,使肾SNS活性降低,可部分地逆转HRS患者的肾功能衰竭[8]。表明SNS作为重要的神经体液因素参与了HRS的发病机制。
3.精氨酸血管加压素(AVP)。AVP的合成位于下丘脑视上核与室旁核,受渗透性(血浆渗透压)与非渗透性两大因素调控。AVP的作用由V1、V2二种受体介导,前者又分为V1a、V1b二种。AVP的缩血管作用主要通过分布于血管组织(包括肾血管)的V1a受体介导,而对水的转运则通过位于肾脏集合管的V2受体导。有学者证实[9],肝硬化大鼠下丘脑AVPmRNA水平明显升高,间接表明AVP的合成和分泌处于兴奋状态。而Arroyo等[10]则发现,肝硬化腹水患者AVP的血浆水平是升高的,且与RBF和GFR呈负相关,表明该激素在HRS发病机制中可能发挥作用。
, http://www.100md.com
4.内皮素(ET)。ET是由21个氨基酸组成的肽,它是主要引起肾血管和肾小球系膜细胞收缩的强效激动剂。多数学者证实,晚期肝病患者的循环ET-1的水平是升高的,而升高的多少与肾功能衰竭的程度有关[11]。最近有资料表明,在HRS患者体内急性给予ET-1受体(BQ123)的特异性拮抗剂后,其肾功能得到改善,这就证明了ET作为HRS中的一种血管收缩因子而发挥其重要作用[12]。然而,引起ET血浆浓度升高的原因至今尚不清楚。
5.半胱氨酰白三烯。 白三烯C4和D4由髓样细胞簇的炎性细胞产生,并已证实在内毒素血症、补体活化或各种细胞因子的刺激下由肾脏合成。它们是二个强效肾血管收缩剂并可使离体肾小球系膜细胞收缩。有证据表明,HRS患者体内全身和肾脏局部白三烯C4、D4、E4等由胆汁排泄发生障碍,转而自肾脏排出,故其尿内浓度显著升高。HRS患者体内所测得的白三烯血浆水平太低,以致不能对肾循环起直接效应,但其肾脏合成的白三烯可能参与了肾功能的调节[4]。
, 百拇医药
6.腺苷。腺苷引起内脏血管扩张,但却能使肾血管收缩。研究证实,肝硬化患者给予潘生丁,通过抑制摄取和降解使内源性腺苷水平升高,进而导致RBF减少[13]。
7.内毒素。内毒素是一个强效肾血管收缩剂,肝硬化患者在受到严重的细菌感染后可进展为HRS[14]。有人观察给清醒大鼠注射内毒素,引起GFR和尿量明显减少,而血压并未降低,表明内毒素可直接影响肾功能。也有人认为内毒素可刺激血抗素生成增多,后者与HRS发生有密切关系。
8.血栓素A2(TXA2)。TXA2由肾脏局部缺血刺激产生,它可导制血管的肾小球系膜细胞收缩。近年来发现,HRS患者尿中前列腺素E2减少的同时TXA2水平明显增高,而不伴有HRS的肝硬化腹水患者则无此作用,推测TXA2在HRS发病机制中有重要作用。但另有资料表明,用血栓素合成酶抑制剂——达唑氧苯(Dazoxiben)抑制TXA2的合成并不能改善肾功能[15],表明它在HRS发病机制中可能不是一个主要的血管收缩因子。
, 百拇医药
综上所述,HRS所涉及的肾血管收缩因素可归纳为两大类。一类是由于肝功能严重障碍致使肝脏不能清除循环有毒物质所产生的,如内毒素等;另一类则是由于低血容量和门脉高压导致有效循环血量减少而产生的,它们通过RAAS和SNS兴奋性增强及其它血管活性物质的综合作用使肾血管持续收缩。
(二)血管扩张因子
许多有关肝硬化病人或实验动物的研究表明,体内的肾血管扩张因子通过对肾循环的保护(因血管收缩因子具有不利效应),在维持肾灌注方面起决定性的作用。
1.前列腺素(PGs)。PGs由花生四烯酸经环氧合酶、过氧化物酶等催化生成,其形式多样,并广泛存在于机体组织中,它是肾内最重要的血管扩张因子,能够抵消因血管收缩因子水平的升高而在肾功能方面产生的效应,同时又不破坏其全身作用。业已证实,非甾体抗炎药物(NSAIDS)如消炎痛可抑制肾脏合成PGs。有人观察肝硬化腹水病人给予NSAIDS(即使仅给一次剂量)可导制RBF和GFR的显著降低,表明肾脏PGs在肝硬化腹水肾灌注的维持中起重要作用[16]。前列环素(PGI2)是在血管内膜合成的一个全身性血管扩张剂,它可因内脏小动脉的剪切力刺激而引起分泌。实验证明,在失代偿性肝硬化中,PGI2的全身和肾脏代谢产物的尿排泄是增加的,且其血浆水平也可能升高。而给予环氧合酶抑制剂能显著地增加意识清醒的肝硬化腹水大鼠的肾血管阻力和减少RBF,表明实验型肝硬化腹水大鼠肾脏PGI2合成的增加是维持肾灌注的代偿机制[17]。有学者认为,肝硬化腹水患者在不伴肾功能衰竭时,RAAS和SNS的活性及其它血管收缩因子水平虽然增高,但因肾脏合成PGs增多,肾灌注仍可维持,而严重肝病进展至HRS时,其RBF的GFR下降可能是由于肾血管收缩增强而肾脏合成PGs不足的结果。
, http://www.100md.com
2.一氧化氮(NO)。NO是由L-精氨酸经一氧化氮合酶(NOS)催化生成的无机小分子化合物。它在体内广泛分布,参与多种生理和病理学效应。近几年来,人们日益清楚地认识到,NO作为一种引起肝硬化全身和肾脏改变的介质起着非常重要的作用,它可能是参与维持肝硬化肾灌注的血管舒张因子之一。有资料表明,自胆管结扎肝硬化大鼠分离的肾小球的基础NO量是增加的,而引起NO产生增加的主要原因则是诱导性NOS(iNOS)的诱导激活[18]。Bosch等[19]也证明,用四氯化碳诱导的肝硬化腹水大鼠肾脏NOSⅢ蛋白表达是增加的,并通过免疫组织化学方法证实它主要位于肾小动脉内皮。最近的研究还揭示,实验性肝硬化大鼠表现为内皮依赖性肾血管扩张的增强[20]和对NO抑制的肾敏感性增加[17]。当实验性肝硬化腹水动物体内PGs或NO分别被抑制时,肾灌注仅表现为一定程度的降低,而当NO和PGs同时被抑制时则出现显著的肾血管收缩,表明NO和PGs相互作用以维持肝硬化肾脏血流动力学[17]。
, 百拇医药
3.利钠肽。自50年代Kisch首先从豚鼠的心房细胞中发现了新型的体液调节激素——心房利钠肽(ANP)以来,很多学者又相继发现α、β、γ型ANP、BNP、CNP,它们均为ANP家族。随后,日本学者竹井祥郎在鳝鱼心室中提取出了具有不同结构特点的心室利钠肽(VNP)。已知ANP和VNP都具有排钠利尿、增加GFR和降低血压的作用,但VNP较ANP具有更强的活性,目前VNP尚未在人体内发现[21]。实验证明,利钠肽是血管收缩和抑钠尿排泄系统的主要负性调节剂,它在钠潴留状态中对维持GFR和促钠尿排泄起着决定性的作用。最近有人观察到,肝硬化腹水大鼠血浆利钠肽水平是升高的,且增加的利钠肽主要来自心室而不是心房[22]。另有学者用选择性拮抗剂HS-142-1急性阻滞肝硬化腹水大鼠利钠肽A和B受体引起显著的肾血管收缩而不导致全身血流动力学改变,表明利钠肽参与了肝硬化肾灌注的维持[23]。但目前还不清楚这些利钠肽是来自肾外亦或在肾内合成。
综上所述,肾血管扩张因子的活性在肝硬化是增强的,并在肾灌注的调节中,尤其是在肾血管收缩机制过度激活的病理情况下起着关键性的作用。
, 百拇医药
二、发病机制
HRS按其临床特征的不同分为二型。Ⅰ型HRS以短期内(数天或几周)快速而进行性的血尿素氮(BUN)和血肌酐(Cr)的增加为特征,其肾功衰竭常伴随着进行性的尿量减少、显著的钠潴溜和低钠血症。它多发生于具有晚期肝功能衰竭信号如黄疸、脑病和/或凝血病的极严重肝病患者,是肝硬化预后最差的并发症[24]。Ⅱ型HRS以中度而稳定的GFR的下降(BUN和血清Cr常分别低于50mg/dl和2mg/dl)为特征。它常发生于相对地尚保存着肝功能的患者,其主要临床后果是形成对利尿药具有抵抗性的腹水[24]。有人预测,Ⅱ型HRS患者存活时间长于Ⅰ型HRS,但又短于没有肾功能衰竭的肝硬化腹水患者。
虽然HRS按其临床特征的不同分为Ⅰ型和Ⅱ型,但有学者认为它们的发病机制则可能是相同的[24]。目前已经提出了二个解释HRS患者体内出现的显著肾脏低灌注的理论。其一,肾脏低灌注与患病的肝脏本身有关,而与全身血流动力学方面的紊乱没有任何联系;其二,肾脏低灌注在发病机制上与全身血流动力学变化有关,而HRS只是肝硬化动脉充盈不足的外周表现之一。
, http://www.100md.com
直接的肝——肾关系理论主要通过以下二个不同的机制得到证实(图1):第一,HRS体内肝源性肾血管扩张因子合成或释放的减少,引起肾血管收缩,进而导制肾灌注的降低;第二,肝脏可通过肝肾反射来调节肾功能,HRS则可能是肝硬化进展期间肝肾反射持续激活的结果[25]。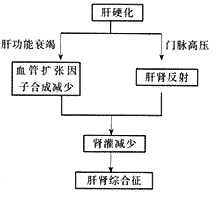
图1 以直接的肝-肾关系理论为基础的肝肾综合征发病机制
第二个理论通常用动脉扩张假说加以阐述(图2)[26]。该假说提出,引起肾脏低灌注的动脉循环的充盈不足可能不是血管内容量减少的结果,而是外周小动脉尤其是内脏循环中小动脉扩张的缘故。正如传统的充盈不足理论所表明的那样,这将导致进行性的压力感受器介导的全身血管收缩因子的激活,进而导致肾循环及其它血管床的血管收缩。内脏循环则由于存在强有力的局部血管扩张因子的刺激,避开了血管收缩因子的作用,而呈持续的、显著的血管扩张。早期随着腹水的进展,尽管血管收缩系统过度激活,但肾灌注通过血管扩张因子合成增加/激活将维持在正常或接近正常水平。而不能被血管扩张因子对抗的全身血管收缩因子的最大限度的激活或血管扩张因子活性的下降和/或肾内血管收缩因子产生的增加,亦或上述二种因素同时存在将导致肾脏低灌注向HRS进展[27]。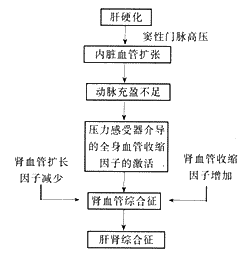
, 百拇医药
图2 动脉扩张假说所示肝肾综合征发病机制
参考文献
1 Koppel MH,C/oburn JN,MimsMM,Goldstein H, B/oyle JD,Rubini HE:Transplantation of cadaveric kidneys from patients with hepa torenal syndrome.Evidence for the functional nature of renal failure in advanced liver disease.N Engl J Med 1969;289∶1155-9.
2 Maroto A,GinèsA,Salo J,Clària J,et al.Diagnosis of functional kidney failure of cirrhosis with Doppler sonography:Prognostic value of resistive inde x.Hepatology 1994;20∶839-844.
, http://www.100md.com
3 Wilkinson SP,Williams R.Rewin-angiotensin-aldosterone system in cirr lnosis.Gut 1980;21∶545-554.
4 K.Moore.The hepatorenal syndrome.Clinical Science 1997;92∶433-443.
5 Hall JE,Brands MW.The renin-angiotensin system:renal mechansuns and c irculatory homestasis.ln:Seldin DW,Giebsch G(eds):The kidney:Physiology and path ophysiology.New york,Raven Press,1992,PP 1455∶1504.
6 Henriksen JH,Ring-Larsen H.Hepatorenal disorders:role of the sympathd tic nervous system.Sem Liver Dis 1994;14∶35-43.
, 百拇医药
7 Henriksen JH,Ring-Larsen H,Christensen NJ.Auronomic nervous function in liver disease.In:130m20n A,Blendis LM,eds.Cardiovasculor complications of liv er disease.Ed 1.Boca Raton:CRC Press,1900∶63-79.
8 Solis-Herruzo JA,Duran A,Favelza V,Castellano G,et al.Effects of lumb ar sympathetic block on kidney functiou in cirrlnotic patients with HRS.J Hepato l 1987;5∶167-73.
9 顾勇,陈靖,马骥,等.下丘脑AVP及肾脏皮质V1a受体mRNA在肝硬化大鼠中的 表达及黄花的作用。中华消 杂志.1998;18∶105-107.
, http://www.100md.com
10 Arroyo V,Gines P,Jimenez W,Rodes J.Ascites,renal failure and electrol yte disorders in cirrhosis.Pathogenesis,diagnosis and treatment,in Oxford Texboo k of clinical Hepatdogy,edited by mcintype N,Benhamou JP,Bircher J,Oxford,Oxford Medical Publications,1991,pp429-470.
11 Epstein M,Goligorsky MS.Endothelin and witric oxide in hepatorenal sy ndrome:a balence reset.J Nephro(1997;10(3):120-35.
12 Soper PR,Latif AB,Bending MR.Amelioration of hepatorenal syndrome wit h seleetive endothelin-A anatagonist.Lancet 1996;347∶1842-1843.
, 百拇医药
13 Lloch J,Gines P,Arroyo V,Salmeron JM,et al.Effect of dipyridamole on kidney function in cirrhosis.Hepatology 1993;17∶59-64.
14 Bourgiognie JJ,Valle GA.Endotoxin and renal failure in Liver Baltimor e,Williams & Wilkins,1988,PP486-507.
15 Zipser RD,Kronborg I,Rector W,Reynolds T,et al.Therapeutic trial of t hromboxane synthesis inlibition in the HRS.Gastroenterlolgy 1984;87∶1228-1232 .
16 Quintero E,Ginès P,Arroyo V,et al.Sulindac reduces the urinary excre tion of prostaglandins and impairs renal function in cirrhosis with ascites.Neph ron 1986;42∶298-303.
, http://www.100md.com
17 Ros J,Clària J,Jiménez W,Bosch-Marcé M,et al.Role of uitric oxide and prostacyclin in the control of renal perfusion in experimental cirrhosis.He patology 1995;22∶915-920.
18 Griado M,Flores O,Ortiz M.C,Hidalgo F,et al.Elevated glounerular and blood Mononuclear lymphocyte witric oxide production in rats with clrouic bile d uet ligation:Role of inducible witric oxide synthase activation.Hepatology 1997; 26∶267-276.
, 百拇医药
19 Basch-Marcé M,Morales-Ruiz M,Jiménez W,Bordas N,et al.Increased r enal expression of witric oxide synthase type Ⅲ in cirrhotic rats with ascites. Hepatology 1998;27∶1191-1199.
20 Garcia-Estan J,Atucha NM,Sabio JM,Vargas F,et al.Increased endotheli um-dependent renal vasodilation in cirrhotic rats.Am J Physiol 1994;267∶R549- R553.
21 刘玉福,赵作光,高风和,等.生理剂量下VNP与ANP的生理作用比较.白求恩 医科大学学报,1997;23∶270-271.
, http://www.100md.com
22 Martic P-y,Ohara M,Gines P,Ding LX,et al.Nitric oxide synthase (NOS) inhibition for one week improves renal sodium and water excretion in cirrhotic rats with ascites.J.Clin.Invest,1998;101∶235-242.
23 Angeli P,Jiménez W,Arroyo V,et al.Renal effects of natriuretic popti de receptor blockade in cirrhotic rats with ascites.Hepatology 1994;20∶948-954 .
24 Ginès A,Escorsell A,Ginès P,et al.Incidence,predicsive factors,and prognosis of the hepatorenal syndrome in cirrhosis with ascites.Gastroenterology 1993;105∶229-236.
, 百拇医药
25 Lang F,Gerok W,Hussiger D:New clues to the patho physiology of hepa torenal failure.Clin Investig(Germany) 1993;71∶93-97.
26 Schrier RW,Arroyo V,Bernardi M,et al.Peripheral arterial vasodilation hypothesis:A proposal for the initiation of renal sodium and water retention in cirrhosis.Hepatology 1988;8∶1151-1157.
27 Bataller R,Sort P,Ginès P,and Arroyo V.Hepatorenal syndrome:Definiti on,Pathophysiology,clinical features and management.Kidney International 1998;53 (Sup.66)∶pp.S47-S53., 百拇医药
单位:谭凯 王天才 430030同济医科大学附属同济医院
关键词:
德国医学/990520 肝肾综合征(HRS)是慢性肝病和晚期肝功能衰竭患者的一个常见并发症,它以肾功能衰竭和显著的全身血流动力学异常以及内源性血管活性系统的改变为特征。
晚期肝病患者可发生肾功能衰竭,这在19世纪末就已得到公认。但直到50年代末才出现了详细描述这一综合征的报道。有关HRS理论基础的主要论断是:肾功能衰竭常呈急进性发展并与全身循环的减少有关;尽管肾功能衰竭严重,但这些患者的肾脏没有显著的组织学异常,HRS患者的肾脏移植给慢性肾功能衰竭患者时可使其肾功能恢复正常,而进行肝移植后的HRS患者其肾功能衰竭能逆转[1]。然而,有关HRS的发病机制至今尚未完全阐明,有些观点亦存在较大争议。本文就HRS的病理生理学及其发病机制作一简述。
, 百拇医药
一、病理生理学
显著的肾血管收缩导致肾脏低灌注是HRS的特点。HRS患者体内存在的显著的肾血管收缩已经通过许多检测方法得到证实,包括肾动脉造影术、133氙洗出术、对氨马脲酸清除率及最近应用的多普勒超声检查。应用这些技术已经证明了肝硬化腹水患者肾灌注的变化是连续的,而HRS则是这一系列变化的终点。与在肾血管系统中发现的显著的血管收缩相反,肾外血流动力学以低全身血管阻力和动脉低血压为特征[2]。
(一)血管收缩因子
导致HRS肾血管收缩的潜在介质已经涉及到肾素——血管紧张素——醛固酮系统、交感神经系统和在肾循环中具有血管收缩效应的一些主要系统。这些血管收缩系统的活性在肝硬化腹水病人,尤其在并发HRS的患者是增加的。
1.肾素——血管紧张素——醛固酮系统(RAAS)。Wilkinson等[3]证实,50%~80%的失代偿性肝硬化患者的RAAS活性是增强的,且在HRS患者其活性进一步提高,表明血管紧张素Ⅱ(ATⅡ)在HRS肾血管收缩的发病机制中起作用。有资料表明,用肌丙抗增压素成血管紧张素转化酶抑制剂抑制肝硬化RAAS引起显著的低血压和肾小球滤过率(GFR)的下降,相反,肝硬化患者在灌注ATⅡ后其GFR增加,这可能是ATⅡ导致动脉压增加的结果[4]。因此,认为RAAS对肾功能的影响可能与全身血流动力学变化有关。但另有资料表明,ATⅡ也可产生于肾脏循环内[5],所以肾素—血管紧张素系统亦可能作为局部血管收缩因子参与肝硬化肾血管收缩的发病机制。
, 百拇医药
2.交感神经系统(SNS)。 自80年代以来,人们已经认识到了肝肾神经支配的重要性。研究证实,HRS患者的SNS高度激活[6],其动脉和肾静脉的去甲肾上腺素(NE)浓度与肾血浆流量(RBF)呈负相关[7]。肾脏SNS的激活通过入球小动脉的收缩,导制RBF和GFR的减少及钠潴留。相反,麻醉阻滞腰SNS,使肾SNS活性降低,可部分地逆转HRS患者的肾功能衰竭[8]。表明SNS作为重要的神经体液因素参与了HRS的发病机制。
3.精氨酸血管加压素(AVP)。AVP的合成位于下丘脑视上核与室旁核,受渗透性(血浆渗透压)与非渗透性两大因素调控。AVP的作用由V1、V2二种受体介导,前者又分为V1a、V1b二种。AVP的缩血管作用主要通过分布于血管组织(包括肾血管)的V1a受体介导,而对水的转运则通过位于肾脏集合管的V2受体导。有学者证实[9],肝硬化大鼠下丘脑AVPmRNA水平明显升高,间接表明AVP的合成和分泌处于兴奋状态。而Arroyo等[10]则发现,肝硬化腹水患者AVP的血浆水平是升高的,且与RBF和GFR呈负相关,表明该激素在HRS发病机制中可能发挥作用。
, http://www.100md.com
4.内皮素(ET)。ET是由21个氨基酸组成的肽,它是主要引起肾血管和肾小球系膜细胞收缩的强效激动剂。多数学者证实,晚期肝病患者的循环ET-1的水平是升高的,而升高的多少与肾功能衰竭的程度有关[11]。最近有资料表明,在HRS患者体内急性给予ET-1受体(BQ123)的特异性拮抗剂后,其肾功能得到改善,这就证明了ET作为HRS中的一种血管收缩因子而发挥其重要作用[12]。然而,引起ET血浆浓度升高的原因至今尚不清楚。
5.半胱氨酰白三烯。 白三烯C4和D4由髓样细胞簇的炎性细胞产生,并已证实在内毒素血症、补体活化或各种细胞因子的刺激下由肾脏合成。它们是二个强效肾血管收缩剂并可使离体肾小球系膜细胞收缩。有证据表明,HRS患者体内全身和肾脏局部白三烯C4、D4、E4等由胆汁排泄发生障碍,转而自肾脏排出,故其尿内浓度显著升高。HRS患者体内所测得的白三烯血浆水平太低,以致不能对肾循环起直接效应,但其肾脏合成的白三烯可能参与了肾功能的调节[4]。
, 百拇医药
6.腺苷。腺苷引起内脏血管扩张,但却能使肾血管收缩。研究证实,肝硬化患者给予潘生丁,通过抑制摄取和降解使内源性腺苷水平升高,进而导致RBF减少[13]。
7.内毒素。内毒素是一个强效肾血管收缩剂,肝硬化患者在受到严重的细菌感染后可进展为HRS[14]。有人观察给清醒大鼠注射内毒素,引起GFR和尿量明显减少,而血压并未降低,表明内毒素可直接影响肾功能。也有人认为内毒素可刺激血抗素生成增多,后者与HRS发生有密切关系。
8.血栓素A2(TXA2)。TXA2由肾脏局部缺血刺激产生,它可导制血管的肾小球系膜细胞收缩。近年来发现,HRS患者尿中前列腺素E2减少的同时TXA2水平明显增高,而不伴有HRS的肝硬化腹水患者则无此作用,推测TXA2在HRS发病机制中有重要作用。但另有资料表明,用血栓素合成酶抑制剂——达唑氧苯(Dazoxiben)抑制TXA2的合成并不能改善肾功能[15],表明它在HRS发病机制中可能不是一个主要的血管收缩因子。
, 百拇医药
综上所述,HRS所涉及的肾血管收缩因素可归纳为两大类。一类是由于肝功能严重障碍致使肝脏不能清除循环有毒物质所产生的,如内毒素等;另一类则是由于低血容量和门脉高压导致有效循环血量减少而产生的,它们通过RAAS和SNS兴奋性增强及其它血管活性物质的综合作用使肾血管持续收缩。
(二)血管扩张因子
许多有关肝硬化病人或实验动物的研究表明,体内的肾血管扩张因子通过对肾循环的保护(因血管收缩因子具有不利效应),在维持肾灌注方面起决定性的作用。
1.前列腺素(PGs)。PGs由花生四烯酸经环氧合酶、过氧化物酶等催化生成,其形式多样,并广泛存在于机体组织中,它是肾内最重要的血管扩张因子,能够抵消因血管收缩因子水平的升高而在肾功能方面产生的效应,同时又不破坏其全身作用。业已证实,非甾体抗炎药物(NSAIDS)如消炎痛可抑制肾脏合成PGs。有人观察肝硬化腹水病人给予NSAIDS(即使仅给一次剂量)可导制RBF和GFR的显著降低,表明肾脏PGs在肝硬化腹水肾灌注的维持中起重要作用[16]。前列环素(PGI2)是在血管内膜合成的一个全身性血管扩张剂,它可因内脏小动脉的剪切力刺激而引起分泌。实验证明,在失代偿性肝硬化中,PGI2的全身和肾脏代谢产物的尿排泄是增加的,且其血浆水平也可能升高。而给予环氧合酶抑制剂能显著地增加意识清醒的肝硬化腹水大鼠的肾血管阻力和减少RBF,表明实验型肝硬化腹水大鼠肾脏PGI2合成的增加是维持肾灌注的代偿机制[17]。有学者认为,肝硬化腹水患者在不伴肾功能衰竭时,RAAS和SNS的活性及其它血管收缩因子水平虽然增高,但因肾脏合成PGs增多,肾灌注仍可维持,而严重肝病进展至HRS时,其RBF的GFR下降可能是由于肾血管收缩增强而肾脏合成PGs不足的结果。
, http://www.100md.com
2.一氧化氮(NO)。NO是由L-精氨酸经一氧化氮合酶(NOS)催化生成的无机小分子化合物。它在体内广泛分布,参与多种生理和病理学效应。近几年来,人们日益清楚地认识到,NO作为一种引起肝硬化全身和肾脏改变的介质起着非常重要的作用,它可能是参与维持肝硬化肾灌注的血管舒张因子之一。有资料表明,自胆管结扎肝硬化大鼠分离的肾小球的基础NO量是增加的,而引起NO产生增加的主要原因则是诱导性NOS(iNOS)的诱导激活[18]。Bosch等[19]也证明,用四氯化碳诱导的肝硬化腹水大鼠肾脏NOSⅢ蛋白表达是增加的,并通过免疫组织化学方法证实它主要位于肾小动脉内皮。最近的研究还揭示,实验性肝硬化大鼠表现为内皮依赖性肾血管扩张的增强[20]和对NO抑制的肾敏感性增加[17]。当实验性肝硬化腹水动物体内PGs或NO分别被抑制时,肾灌注仅表现为一定程度的降低,而当NO和PGs同时被抑制时则出现显著的肾血管收缩,表明NO和PGs相互作用以维持肝硬化肾脏血流动力学[17]。
, 百拇医药
3.利钠肽。自50年代Kisch首先从豚鼠的心房细胞中发现了新型的体液调节激素——心房利钠肽(ANP)以来,很多学者又相继发现α、β、γ型ANP、BNP、CNP,它们均为ANP家族。随后,日本学者竹井祥郎在鳝鱼心室中提取出了具有不同结构特点的心室利钠肽(VNP)。已知ANP和VNP都具有排钠利尿、增加GFR和降低血压的作用,但VNP较ANP具有更强的活性,目前VNP尚未在人体内发现[21]。实验证明,利钠肽是血管收缩和抑钠尿排泄系统的主要负性调节剂,它在钠潴留状态中对维持GFR和促钠尿排泄起着决定性的作用。最近有人观察到,肝硬化腹水大鼠血浆利钠肽水平是升高的,且增加的利钠肽主要来自心室而不是心房[22]。另有学者用选择性拮抗剂HS-142-1急性阻滞肝硬化腹水大鼠利钠肽A和B受体引起显著的肾血管收缩而不导致全身血流动力学改变,表明利钠肽参与了肝硬化肾灌注的维持[23]。但目前还不清楚这些利钠肽是来自肾外亦或在肾内合成。
综上所述,肾血管扩张因子的活性在肝硬化是增强的,并在肾灌注的调节中,尤其是在肾血管收缩机制过度激活的病理情况下起着关键性的作用。
, 百拇医药
二、发病机制
HRS按其临床特征的不同分为二型。Ⅰ型HRS以短期内(数天或几周)快速而进行性的血尿素氮(BUN)和血肌酐(Cr)的增加为特征,其肾功衰竭常伴随着进行性的尿量减少、显著的钠潴溜和低钠血症。它多发生于具有晚期肝功能衰竭信号如黄疸、脑病和/或凝血病的极严重肝病患者,是肝硬化预后最差的并发症[24]。Ⅱ型HRS以中度而稳定的GFR的下降(BUN和血清Cr常分别低于50mg/dl和2mg/dl)为特征。它常发生于相对地尚保存着肝功能的患者,其主要临床后果是形成对利尿药具有抵抗性的腹水[24]。有人预测,Ⅱ型HRS患者存活时间长于Ⅰ型HRS,但又短于没有肾功能衰竭的肝硬化腹水患者。
虽然HRS按其临床特征的不同分为Ⅰ型和Ⅱ型,但有学者认为它们的发病机制则可能是相同的[24]。目前已经提出了二个解释HRS患者体内出现的显著肾脏低灌注的理论。其一,肾脏低灌注与患病的肝脏本身有关,而与全身血流动力学方面的紊乱没有任何联系;其二,肾脏低灌注在发病机制上与全身血流动力学变化有关,而HRS只是肝硬化动脉充盈不足的外周表现之一。
, http://www.100md.com
直接的肝——肾关系理论主要通过以下二个不同的机制得到证实(图1):第一,HRS体内肝源性肾血管扩张因子合成或释放的减少,引起肾血管收缩,进而导制肾灌注的降低;第二,肝脏可通过肝肾反射来调节肾功能,HRS则可能是肝硬化进展期间肝肾反射持续激活的结果[25]。
图1 以直接的肝-肾关系理论为基础的肝肾综合征发病机制
第二个理论通常用动脉扩张假说加以阐述(图2)[26]。该假说提出,引起肾脏低灌注的动脉循环的充盈不足可能不是血管内容量减少的结果,而是外周小动脉尤其是内脏循环中小动脉扩张的缘故。正如传统的充盈不足理论所表明的那样,这将导致进行性的压力感受器介导的全身血管收缩因子的激活,进而导致肾循环及其它血管床的血管收缩。内脏循环则由于存在强有力的局部血管扩张因子的刺激,避开了血管收缩因子的作用,而呈持续的、显著的血管扩张。早期随着腹水的进展,尽管血管收缩系统过度激活,但肾灌注通过血管扩张因子合成增加/激活将维持在正常或接近正常水平。而不能被血管扩张因子对抗的全身血管收缩因子的最大限度的激活或血管扩张因子活性的下降和/或肾内血管收缩因子产生的增加,亦或上述二种因素同时存在将导致肾脏低灌注向HRS进展[27]。
, 百拇医药
图2 动脉扩张假说所示肝肾综合征发病机制
参考文献
1 Koppel MH,C/oburn JN,MimsMM,Goldstein H, B/oyle JD,Rubini HE:Transplantation of cadaveric kidneys from patients with hepa torenal syndrome.Evidence for the functional nature of renal failure in advanced liver disease.N Engl J Med 1969;289∶1155-9.
2 Maroto A,GinèsA,Salo J,Clària J,et al.Diagnosis of functional kidney failure of cirrhosis with Doppler sonography:Prognostic value of resistive inde x.Hepatology 1994;20∶839-844.
, http://www.100md.com
3 Wilkinson SP,Williams R.Rewin-angiotensin-aldosterone system in cirr lnosis.Gut 1980;21∶545-554.
4 K.Moore.The hepatorenal syndrome.Clinical Science 1997;92∶433-443.
5 Hall JE,Brands MW.The renin-angiotensin system:renal mechansuns and c irculatory homestasis.ln:Seldin DW,Giebsch G(eds):The kidney:Physiology and path ophysiology.New york,Raven Press,1992,PP 1455∶1504.
6 Henriksen JH,Ring-Larsen H.Hepatorenal disorders:role of the sympathd tic nervous system.Sem Liver Dis 1994;14∶35-43.
, 百拇医药
7 Henriksen JH,Ring-Larsen H,Christensen NJ.Auronomic nervous function in liver disease.In:130m20n A,Blendis LM,eds.Cardiovasculor complications of liv er disease.Ed 1.Boca Raton:CRC Press,1900∶63-79.
8 Solis-Herruzo JA,Duran A,Favelza V,Castellano G,et al.Effects of lumb ar sympathetic block on kidney functiou in cirrlnotic patients with HRS.J Hepato l 1987;5∶167-73.
9 顾勇,陈靖,马骥,等.下丘脑AVP及肾脏皮质V1a受体mRNA在肝硬化大鼠中的 表达及黄花的作用。中华消 杂志.1998;18∶105-107.
, http://www.100md.com
10 Arroyo V,Gines P,Jimenez W,Rodes J.Ascites,renal failure and electrol yte disorders in cirrhosis.Pathogenesis,diagnosis and treatment,in Oxford Texboo k of clinical Hepatdogy,edited by mcintype N,Benhamou JP,Bircher J,Oxford,Oxford Medical Publications,1991,pp429-470.
11 Epstein M,Goligorsky MS.Endothelin and witric oxide in hepatorenal sy ndrome:a balence reset.J Nephro(1997;10(3):120-35.
12 Soper PR,Latif AB,Bending MR.Amelioration of hepatorenal syndrome wit h seleetive endothelin-A anatagonist.Lancet 1996;347∶1842-1843.
, 百拇医药
13 Lloch J,Gines P,Arroyo V,Salmeron JM,et al.Effect of dipyridamole on kidney function in cirrhosis.Hepatology 1993;17∶59-64.
14 Bourgiognie JJ,Valle GA.Endotoxin and renal failure in Liver Baltimor e,Williams & Wilkins,1988,PP486-507.
15 Zipser RD,Kronborg I,Rector W,Reynolds T,et al.Therapeutic trial of t hromboxane synthesis inlibition in the HRS.Gastroenterlolgy 1984;87∶1228-1232 .
16 Quintero E,Ginès P,Arroyo V,et al.Sulindac reduces the urinary excre tion of prostaglandins and impairs renal function in cirrhosis with ascites.Neph ron 1986;42∶298-303.
, http://www.100md.com
17 Ros J,Clària J,Jiménez W,Bosch-Marcé M,et al.Role of uitric oxide and prostacyclin in the control of renal perfusion in experimental cirrhosis.He patology 1995;22∶915-920.
18 Griado M,Flores O,Ortiz M.C,Hidalgo F,et al.Elevated glounerular and blood Mononuclear lymphocyte witric oxide production in rats with clrouic bile d uet ligation:Role of inducible witric oxide synthase activation.Hepatology 1997; 26∶267-276.
, 百拇医药
19 Basch-Marcé M,Morales-Ruiz M,Jiménez W,Bordas N,et al.Increased r enal expression of witric oxide synthase type Ⅲ in cirrhotic rats with ascites. Hepatology 1998;27∶1191-1199.
20 Garcia-Estan J,Atucha NM,Sabio JM,Vargas F,et al.Increased endotheli um-dependent renal vasodilation in cirrhotic rats.Am J Physiol 1994;267∶R549- R553.
21 刘玉福,赵作光,高风和,等.生理剂量下VNP与ANP的生理作用比较.白求恩 医科大学学报,1997;23∶270-271.
, http://www.100md.com
22 Martic P-y,Ohara M,Gines P,Ding LX,et al.Nitric oxide synthase (NOS) inhibition for one week improves renal sodium and water excretion in cirrhotic rats with ascites.J.Clin.Invest,1998;101∶235-242.
23 Angeli P,Jiménez W,Arroyo V,et al.Renal effects of natriuretic popti de receptor blockade in cirrhotic rats with ascites.Hepatology 1994;20∶948-954 .
24 Ginès A,Escorsell A,Ginès P,et al.Incidence,predicsive factors,and prognosis of the hepatorenal syndrome in cirrhosis with ascites.Gastroenterology 1993;105∶229-236.
, 百拇医药
25 Lang F,Gerok W,Hussiger D:New clues to the patho physiology of hepa torenal failure.Clin Investig(Germany) 1993;71∶93-97.
26 Schrier RW,Arroyo V,Bernardi M,et al.Peripheral arterial vasodilation hypothesis:A proposal for the initiation of renal sodium and water retention in cirrhosis.Hepatology 1988;8∶1151-1157.
27 Bataller R,Sort P,Ginès P,and Arroyo V.Hepatorenal syndrome:Definiti on,Pathophysiology,clinical features and management.Kidney International 1998;53 (Sup.66)∶pp.S47-S53., 百拇医药