HPV16致癌分子机制的研究进展
作者:钟才高 李佩珊
单位:湖南医科大学卫生毒理学教研室,长沙 410078
关键词:
卫生毒理学杂志000220 人乳头瘤病毒(Human papillomaviruses,HPV)是一类分子长度大约为8 kb左右的小分子环状双股DNA病毒。通过DNA序列分析,至今已发现人乳头瘤病毒大约有70多型,不同型HPV常常分布于不同组织的上皮表面,大约有20多型HPV嗜好感染粘膜上皮,特别是生殖道的粘膜上皮[1]。大量分子流行病学研究资料表明,人乳头瘤病毒16型(HPV16)与生殖道肿瘤密切相关,在宫颈癌活检组织中,HPV16的阳性检出率显著高于正常宫颈组织[2]。动物致癌实验发现,HPV16及其E6E7基因可明显诱导小鼠宫颈癌的发生[3]。故普遍认为HPV16是宫颈癌发生的“高危”型人乳头瘤病毒。HPV16既可以其基因片段又可以整个DNA形式整合到宿主细胞基因组中[4]。目前,国内外对HPV16致癌分子机制研究甚多,特别是体外研究,现从HPV16的分子生物学角度就HPV16的分子基础与分子致癌机制等方面综述如下。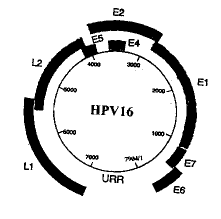
, http://www.100md.com
图1 HPV16基因结构平面示意图
1 HPV16及其基因的分子基础
HPV16是一分子长度为7904 bp的环状双股DNP病毒,1983年从生殖道癌组织细胞中发现了HPV16[5],1985年Seedorf.K等[6]公开发表了HPV16的DNA序列及基因组结构表达产物的生物学作用的研究论文,他们发现HPV16 DNA链第1 bp~60 bp的序列与HPV16al,HPV16b与BPV1一致。HPV16分子结构内含E1、E2、E4、E5、E6、E7早期开放阅读框架(Early open reading frames)和L1,L2晚期开放阅读框架(late open reading frames)。
1.1 HPV16基因组结构[1] HPV16基因组的开放阅读框架内含能产生基因表达产物的早期基因有6个,晚期基因有2个(图1)。早期基因即E1、E2、E4、E5、E6、E7基因的表达产物都控制病毒DNA复制的非结构蛋白,在一些情况下,某些基因与致癌性有关;晚期基因即L1与L2基因的表达产物都是构建病毒壳体的结构蛋白。
, 百拇医药
1.2 HPV16基因及其作用 不同的基因其表达产物有不同的生物学作用(表1)[1],而且在上游控制区(URR)、E6、E7、E1、E2之间有正/负反馈调节环(feedback loop),即E2的基因产物可正/负调节URR中从启动子开始的转录[5]。
表1 HPV16基因及其作用 基因
碱基数
碱基序列
基因表达产物的作用
E1
954 bp
859~2 813
控制病毒DNA的复制
, http://www.100md.com
E2
1127 bp
2 725~3 852
控制转录
E4
287 bp
3 332~3 619
捆绑于细胞角蛋白
E5
236 bp
3 863~4 099
活化生长因子受体
, 百拇医药
E6
494 bp
65~559
捆绑于p53蛋白
E7
314 bp
544~858
捆绑于Rb蛋白
L1
1628 bp
5 526~7 154
构成较大病毒壳体
, http://www.100md.com L2
1523 bp
4 133~5 656
构成较小病毒壳体
URR
813 bp
7 155~7 904/0~64
内含启动子,构成反馈调节环
2 HPV16致癌分子机制研究
2.1 HPV16诱导的体外细胞恶性转化 自1983年从生殖道癌组织细胞中发现HPV16以来,国内外许多学者对HPV16的分子致癌机理进行了广泛研究,其研究结果各异。较早的研究是Sylvie Schneiber-Maunoury等人[7](1987)在角蛋白细胞系SKV中克隆出含有被整合的HPV16 DNA序列的宿主细胞DNA片段。研究结果表明,这种整合在E2与L2之间打断了HPV16 DNA链。并产生一个813碱基对的缺失,从而导致E6与E7的基因表达。在癌前损伤组织细胞就已整合有HPV16序列。Junyile等人[8](1988)研究发现从HPV16阳性的人的肿瘤组织细胞中获得的一段分子长度为4.4 kb的DNA片段(T4.4)(内含HPV16 E6启动子、E6E7、部分E1基因和宿主细胞DNA序列),具有完全转化NIH3T3细胞的活力,并已在转化的细胞内发现有大量的E6E7转录物存在。但是单独的HPV16片段或细胞DNA片段却无这种活性。当用多瘤病毒(polyomavirus)或SV40 DNA的多腺苷酸化序列(片段)取代T4.4中HPV16 DNA片段时,很少或者没有观察到转化活性的恢复。这些结果揭示含有从人肿瘤细胞基因组分离出来的HPV16E6E7 DNA片段具有转化潜力,这种潜力只有在HPV16 DNA被连接到宿主细胞DNA序列中才能实现。细胞DNA序列比以简单提供一个多腺苷酸化DNA片段更为复杂的方式发挥作用。
, http://www.100md.com
为了确定HPV16 DNA分子中那个基因在细胞转化中起作用,Masuo Yutsudo等人[9](1988)构建了含有HPV16早期开放阅读框架(EORF)不同亚基因片段的重组鼠源逆转录病毒的DNA,并测定了它们的恶性转化活性。结果发现E6和E7 ORF是HPV16的转化基因,前者在裸鼠体内决定致瘤性,而后者在软琼脂中影响细胞生长特性(例如细胞饱和密度与克隆形式);并发现在E1-E2 ORF区域可能存在一个抑癌基因,因为含有E1 ORF和E2 ORF片段加E6和E7 ORF片段的重组DNA的致瘤活性明显低于仅仅只有E6和E7 ORF片段DNA。国内张卫等人[10](1991)用HZIP16和HZIP16K 2种质粒,将HPV16的全早期区基因与E6、E7 ORF片段分别转入φ细胞,所产生的重组病毒能诱导NIH3T3细胞发生转化。获得的转化细胞具有接触抑制消失,非锚地依赖性生长,并可使裸鼠致瘤。结果表明,HPV16全EORF DNA片段具有诱导NIH3T3细胞恶性转化的活性。早期区基因中的E6-E7 ORFs也可单独诱导细胞恶性转化。为了探索HPV16长控制区(LCR)在E6E7 ORFs转化中的作用,候芷英等人[11]1994构建了含HPV16 E6E7 ORFs(nt83-855)片段和HPV16长控制区(LCR)加E6E7 ORFs片段(nt7007―7904/0-879)的逆转病毒载体pH21和pH18质粒,利用琼脂转染法,分别将它们导入病毒包装细胞pA317中,经过筛选获得G418抗性的病毒包装细胞,产生的重组病毒H21和H18感染的NIH3T3细胞都具有恶性细胞的形态学特征,并能使裸鼠形成肿瘤。Southern Blot杂交结果表明,上述两个基因片段都被整合到细胞基因组中。细胞转化结果说明,HPV16E6E7基因片段是HPV16转化NIH3T3细胞的关键早期基因,但其自身的LCR在细胞恶性转化过程中并没有显示出重要作用。盖其伟等人[12](1994)在体外用HPV16 DNA完整分子通过磷酸钙沉淀技术转录NIH3T3细胞,从而导致了NIH3T3细胞的恶性转化,并用Southern blot杂交证实HPV16 DNA分子以整合状态存在于NIH3T3细胞DNA中。
, 百拇医药
2.2 HPV16及其基因组与宿主细胞抑癌基因的关系 许多研究表明,HPV16及其基因组的致癌(肿瘤)性或细胞恶性转化作用是由于HPV16的有关基因通过影响宿主细胞抑癌基因表达产物生物活性来实现的。Dyson和Werness分别于1989年、1990年在《Science》杂志上发表论文[13,14],提出HPV16产生的两个病毒蛋白即E6、E7蛋白可能分别捆绑并抑制宿主细胞内的两个关键性的抗癌蛋白即P53蛋白和Rb蛋白。在正常细胞内,P53和Rb调控细胞周期与凋亡,P53与Rb表达产物的失活可导致宿主细胞永生化。E6蛋白可能与P53表达产物结合而导致P53降解失活,继之使含HPV16的宿主细胞生长失控。Magnus Non Knebel Doebejz等人[15](1994)用SW756宫颈癌细胞研究发现:E7蛋白与Rb结合可影响Rb与细胞E2F转录因子的结合,使E2F与Rb分离而具有转录激活功能,从而影响细胞的生长与分化,但在SW756衍生的细胞系中E7的情况并非如此。
, 百拇医药 P16(MTS1)肿瘤抑制基因是一个周期素依赖性激酶(cdk)的抑制基因,它能通过灭活cdk(cdk能使pRb磷酸化)使细胞周期减慢。Nakao-y等人[15]研究发现宫颈上皮细胞被HPV16永生化后,可观察到P16 RNA水平的急剧增加,而且,在随后的永生化过程中能观察到P16蛋白的表达。但是细胞恶性转化后,P16RNA与蛋白水平没有改变。这些结果表明,在恶性转化期间,不会发生P16-cdk-Rb串联体的失活,但在隐藏有HPV16的恶化前期(癌前期)损伤组织的永生化期间,则出现P16-cdk-Rb串联体的失活。
转化生长因子-β1(TGF-β1)是一个细胞分裂因子,对许多上皮细胞、内皮细胞、淋巴细胞等是一潜在性生长抑制剂,而对间质细胞与骨细胞则是促有丝分裂剂。Dey-A等人[17]研究评价了HPV16E6E7癌蛋白对TGF-β1启动子的作用。结果表明,HPV16E6明显诱导TGF-β1启动子活化(6倍于对照组),而HPV16E7则无明显作用。E6作用存在细胞型的特殊性,即仅仅在成纤维细胞中观察到它的作用,在上皮细胞则无。启动子序列分析表明,TGF-β1启动子的-109~-100序列中存在富含有GC的9个碱基对的片断即GGGGCGGGG,它是E6介导的反式活化的主要靶基因,HPV16E6蛋白的123~136位的3个氨基酸对TGF-β1启动子的反式活化至关重要。
, 百拇医药
E-Cadherin是位于染色体16q上,其编码蛋白有细胞表面粘附作用,也属于一种抑癌基因。HPV16使在培养基中的原发角质化细胞永生化,并抑制血清Ca刺激的分化。Wilding-J等人(1996)[18]用一个建立在HPV16基础上的角质化细胞永生化模型分析了ECad-herin的功能与表达的紊乱,也分析了正常角质化细胞所表达的Ca2+-依赖性细胞的细胞粘附分子及其相关蛋白。联合转染HPV16E6和E7所产生的永生化角质化细胞系能降低膜E-Cadherin表达,以及使α、β、γ细胞骨架蛋白(Catenin)从内膜到胞浆重新分布。上皮细胞生长因子受体(EGFr)的过度表达与被HPV16E6E7转染的角质化细胞中E-Cadherin/Catenin复合物的紊乱相关联,可能构成生长调节因子和吸附分子之间功能性相互影响的基础。
2.3 HPV16及其基因在宿主细胞DNA中的整合位点 脆性组氨酸三联体(FHIT)基因是一个被公认的肿瘤抑制基因,其中内含重叠的FRA3B(FRA3B是一个最有活性的染色体断裂点,位于染色体3p14.2处)。HPV16可以频繁地整合到FRA3B部位[19],并发现HPV16与FHIT/FRA3B区域中杂合性丢失有关。
, 百拇医药
Ets家族转录因子erg与ets-2表达变化与宫颈癌的发展有关。二者的表达变化与HPV16永生化的宫颈细胞和宫颈癌细胞系(CX16-2)染色体21q、22.2~22.3的基因改变相联系。而HPV16在CX16-2细胞中染色体21q 22.2~22.3易位断裂点发生整合[20]。
为了弄清楚在宫颈癌细胞中被HPV16整合所破坏的细胞染色体DNA位点的结构特性,Choo-KB等人[21](1996)用PCR技术直接扩增,分析了宫颈癌活检样品中细胞染色体DNA的4个位点。HPV16破坏的位点之一是2号染色体Map-2基因的位置,其他3个破坏位点分别是9号染色体PID,1号染色体PID-2与8号染色体PID-3,以上4对染色体携带着许多从前已测定过的HPV整合部位与染色体脆性部位和Myc癌基因。结果显示,HPV16基因组在宫颈癌细胞DNA中的整合位点多在染色体不稳定部位和具有转录活性的部位。
在许多宫颈肿瘤和从其衍生的细胞系中,HPV16 DNA序列能够整合到宿主细胞基因组中。Bauer-Hofmann-R等人[22](1996)用酶谱分析发现由于病毒DNA的整合大约有7.8 kb的宿主细胞DNA缺失。通过计算机分析,缺失的基因组区段2.3 kb的DNA序列和病毒整合位点上游的1.0 kb的序列与任何已知的人细胞DNA序列无明显同源性。这些发现提示,病毒整合发生在接近被公认的控制序列,病毒与宿主细胞DNA重组不会跟随有整合位点染色质结构上游的重组。RT-PCR实验表明,HPV的特殊转录跨越E2、E4、E5和位于HPV16控制区上游的L1/L2 ORFs。根据这些资料,综合起来考虑在SiHa细胞中,HPV16整合位点上游的宿主细胞DNA区域内含有影响位于HPV16上游控制区(URR)上游的HPV16 ORFs转录的控制元件。
, 百拇医药
3 抗HPV16癌基因的研究
为了评价核酶(Ribozyme)对CV-1细胞中E7基因表达的抑制作用,Huang-Y等人[23](1996)用带有1个降解E7 RNA的核酶和2个加工处理核酶的基因Rz523在5′和3′末端克隆到真核表达质粒PREP9中,在RSV-LTR启动的控制下,通过磷酸钙共沉淀法将其克隆产物质粒PRSV-Rz 523转染到CV-1细胞中。用点杂交法来检测G418抗性细胞中核酶的表达。在CV-1细胞中,稳定表达的核酶在9.0pmol/106细胞水平,其中有活性的核酶分子超过50fmol/106细胞。RNase保护性分析结果表明,在表达核酶CV-1细胞中的E7 RNA片段稳定含量显著地减少了90%。结果表明,用核酶抑制E7基因表达能使宫颈癌恶性表型逆转成为可能。
HPV16E6E7基因在宫颈癌致癌机制中起着中心作用,它们已被公认为与细胞中肿瘤抑制基因p53,p105与Rb等产物有相互作用,为了验证“E6E7在细胞恶性表型的维持中起主动作用”和“E6E7可能是抗基因治疗理论靶点”的假设,Madrigal-M等人[24](1997)用硫代寡脱氧核苷酸(phosphorothioate oligodeoxynucleotides)来攻打宫颈癌细胞系与原发肿瘤外殖体中HPV16E6E7,发现硫代寡脱氧核苷酸具有抗HPV16E6E7产生的增生作用。
, 百拇医药
在探索腺病毒反义RNA转录物对宫颈癌基因治疗可能性的研究中,Hamada-K等人[25](1996)将HPV16E6E7基因的反义RNA转录物通过重组腺病毒载体(Ad5 CMV-HPV16AS)导入隐含有HPV16的宫颈癌细胞中,分析了这些基因的表达对细胞生长与肿瘤生长的影响。Ad5 CMV-HPV16AS的基因框(盒)中含有巨细胞病毒启动子,反义HPV16E6E7基因和SV40多腺苷酸信号。使上述基因框插入到已修改了的腺病毒5的E1缺失区。结果表明,感染有Ad5 CMV-HPV16AS的细胞,其生长明显受到抑制。在体内研究中,给裸鼠注射Ad5 CMV-HPV16AS感染的SiHa细胞完全抑制了鼠的致瘤性。因而,以Ad5 CMV-HPV16AS形式转染含有HPV16E6E7反义RNA细胞,这可能是治疗HPV16阳性宫颈癌的一种具有潜力的新途径。
4 小结
综上所述,HPV16感染宿主细胞后,既可以HPV16全分子,也可以基因片段整合到宿主细胞DNA中(或染色体中),其表达蛋白可通过影响宿主细胞的一些抑癌(肿瘤)基因和相关细胞生长调控因子的生物活性来使被感染的宿主细胞恶性转化或发生癌变。目前普遍认为HPV16分子中E6E7基因是HPV16的致癌基因,并在裸鼠致瘤性实验中得到证实。通过抗HPV16癌基因的体外研究成果,由HPV16引起的宫颈癌的基因治疗存在广阔前景。
, 百拇医药
参考文献
[1] H.D.L.Birley.Human papillomaviruses,cervical cancer and the developing world.Annals of tropical medicine and parasitology,1995,89:453-463.
[2] 蔡红,姚,陈伟丽,等.多聚酶链反应检测宫颈癌HPV感染及分型的研究.中国病毒学,1994,9:291-296.
[3] 钟才高,Donald D.Anthony.HPV16及其基因诱导小鼠宫颈癌的毒理学研究.实用预防医学,1999,6:401-404.
[4] Lazo PA.The molecular genetics of cervical carcinoma.Br J cancer,1999,80:2008-2018.
, 百拇医药
[5] Christopher P Crum,Shannon Barber,James K Roche.Pathobiology of papillomavirusrelated cervical diseases:Prospects for immunodiagnosis.Clinical Microbiology Reviews,1991,4:270-285.
[6] Seedorf K,Kraemmer C,Duerst M,et al.Human papillomavirus type 16 DNA sequence.Virology,1985,145:181-185.
[7] Sylvie schneider-Maunoury,Odile croissant,Gerard Orth.Intergration of human papillomavirus type 16 DNA sequence:a possible early event in the progression of genital tumors.Journal of virology,1987,61:3295-3298.
, 百拇医药
[8] Jun-Yile,Vittorio Defendi.A viral-cellular junction fragment from a human papillomavirus type 16-positive tumour is competent in transformation of NIH3T3 cells.Journal of Virology,1988,62:4420-4426.
[9] Masuo Yutsudo,Yuji Okamoto,Akira Hakura.Functional dissociation of transforming genes of human papillomavirus type 16.Virology,1988,166:594-597.
[10] 张卫,司静懿,李昆,等.人乳头瘤病毒16型亚基因DNA体外转化功能的细胞学研究.病毒学报,1991,7:222-225.
, 百拇医药
[11] 候芷英,孙亚洲,商铭,等.人乳头瘤病毒16型E6E7 ORF转化作用的研究.中华实验和临床病毒学杂志,1994,8:112-115.
[12] 盖其伟,赫翠珍,姚庆英,等.人乳头瘤病毒16型DNA诱导NIH3T3细胞的恶性转化.首都医学院学报,1994,15:109-111.
[13] Dyson N,Howley PM,Munger K,et al.The human papillomavirus 16 E7 oncoprotein is able to bind the retinoblastoma gene product.Science,1989,243:934-936.
[14] Verness BA,Levine AJ,Howley PM.Association of human papillomavirus type 16 and 18 E6 proteins with p53.Science,1990,248:76-79.
, 百拇医药
[15] Magnus von knebel doeberitz,Claudia rittmuller,Frank Aengeneyndt,et al.Reversible repression of papillomavirus oncogene expression in cervical carcinoma cells:Consequences for the phenotype and E6-p53 and E7-pRb interactions.J Virol,1994,68:2811-2821.
[16] Nakao Y,Yang X,Yokoyama M,et al.Induction of p16 during immortalization by HPV16 and 18 and not during malignant transformation.Br J cancer,1997,75:1410-1416.
[17] Dey A,Atcha IA,Bagchi S.HPV16E6 oncoprotein stimulates the transforming growth factor-beta 1 promoter in fibroblasts through a specific GC-rich sequence.virology,1997,228:190-199.
, 百拇医药
[18] Wilding J,Vousden KH,Soutter WP,et al.E-cadherin transfection down-requlates the epidermal growth factor receptor and reverses the invasive phenotype of human papillomavirus-trasfected keratinocytes.Cancer Res,1996,56:5285-5292.
[19] Muller CY,O'Boyle Jd,Fong KM,et al.Abnormalities of fragile histidine triad genomic and complementary DNA in cervical cancer:association with human papillomavirus type. J Natl Cancer Inst,1998,90:433-439.
, 百拇医药
[20] Simpson S,Woodworth CD,Dipaolo JA.Altered expression of Erg and Ets-2 transcription factors is association with genetic changes at 21q22.2-22.3 in immortal and cervical carcinoma cell lines.Oncogene,1997,14(18):2149-2157.
[21] Choo KB,Chen CM,Han CP,et al.Molecular analysis of cellular loci disrupted by papillomavirus 16 intergration in cervical cancer:frequent viral intergration in topologically destabilized and transcriptionally active chromosomal regions.J Med Virol,1996,49:15-22.
, 百拇医药
[22] Bauer Hofmann R,Barghouts C,Auvinen E,et al.Genomic cloning and characterization of the nonoccupied allele correspounding to the intergration site of human papillomavirus type 16 DNA in the cervical cancer cell line SiHa.Virology,1996,217:33-41.
[23] Huang Y,Yong Y,Wang Y,et al.Stable expression of anti-HPV16E7-ribozyme in CV-1 Cell lines.Chin J Biotechnol,1996,12:215-220.
[24] Madrigal M,Janicak MF,Sevin BU,et al.In Vitro antigen therapy targeting HPV16E6 and E7 cervical carcinoma.Gynecol Oncol,1997,64:18-25.
[25] Hamada K,Sakaue M,Alemany R,et al.Adenovirus Mediated transfer of HPV16E6/E7 anti-sense RNA to human cervical cancer cells.Gynecol Oncol,1996,63:219-227.
(收稿日期:1999-10-18), http://www.100md.com
单位:湖南医科大学卫生毒理学教研室,长沙 410078
关键词:
卫生毒理学杂志000220 人乳头瘤病毒(Human papillomaviruses,HPV)是一类分子长度大约为8 kb左右的小分子环状双股DNA病毒。通过DNA序列分析,至今已发现人乳头瘤病毒大约有70多型,不同型HPV常常分布于不同组织的上皮表面,大约有20多型HPV嗜好感染粘膜上皮,特别是生殖道的粘膜上皮[1]。大量分子流行病学研究资料表明,人乳头瘤病毒16型(HPV16)与生殖道肿瘤密切相关,在宫颈癌活检组织中,HPV16的阳性检出率显著高于正常宫颈组织[2]。动物致癌实验发现,HPV16及其E6E7基因可明显诱导小鼠宫颈癌的发生[3]。故普遍认为HPV16是宫颈癌发生的“高危”型人乳头瘤病毒。HPV16既可以其基因片段又可以整个DNA形式整合到宿主细胞基因组中[4]。目前,国内外对HPV16致癌分子机制研究甚多,特别是体外研究,现从HPV16的分子生物学角度就HPV16的分子基础与分子致癌机制等方面综述如下。
, http://www.100md.com
图1 HPV16基因结构平面示意图
1 HPV16及其基因的分子基础
HPV16是一分子长度为7904 bp的环状双股DNP病毒,1983年从生殖道癌组织细胞中发现了HPV16[5],1985年Seedorf.K等[6]公开发表了HPV16的DNA序列及基因组结构表达产物的生物学作用的研究论文,他们发现HPV16 DNA链第1 bp~60 bp的序列与HPV16al,HPV16b与BPV1一致。HPV16分子结构内含E1、E2、E4、E5、E6、E7早期开放阅读框架(Early open reading frames)和L1,L2晚期开放阅读框架(late open reading frames)。
1.1 HPV16基因组结构[1] HPV16基因组的开放阅读框架内含能产生基因表达产物的早期基因有6个,晚期基因有2个(图1)。早期基因即E1、E2、E4、E5、E6、E7基因的表达产物都控制病毒DNA复制的非结构蛋白,在一些情况下,某些基因与致癌性有关;晚期基因即L1与L2基因的表达产物都是构建病毒壳体的结构蛋白。
, 百拇医药
1.2 HPV16基因及其作用 不同的基因其表达产物有不同的生物学作用(表1)[1],而且在上游控制区(URR)、E6、E7、E1、E2之间有正/负反馈调节环(feedback loop),即E2的基因产物可正/负调节URR中从启动子开始的转录[5]。
表1 HPV16基因及其作用 基因
碱基数
碱基序列
基因表达产物的作用
E1
954 bp
859~2 813
控制病毒DNA的复制
, http://www.100md.com
E2
1127 bp
2 725~3 852
控制转录
E4
287 bp
3 332~3 619
捆绑于细胞角蛋白
E5
236 bp
3 863~4 099
活化生长因子受体
, 百拇医药
E6
494 bp
65~559
捆绑于p53蛋白
E7
314 bp
544~858
捆绑于Rb蛋白
L1
1628 bp
5 526~7 154
构成较大病毒壳体
, http://www.100md.com L2
1523 bp
4 133~5 656
构成较小病毒壳体
URR
813 bp
7 155~7 904/0~64
内含启动子,构成反馈调节环
2 HPV16致癌分子机制研究
2.1 HPV16诱导的体外细胞恶性转化 自1983年从生殖道癌组织细胞中发现HPV16以来,国内外许多学者对HPV16的分子致癌机理进行了广泛研究,其研究结果各异。较早的研究是Sylvie Schneiber-Maunoury等人[7](1987)在角蛋白细胞系SKV中克隆出含有被整合的HPV16 DNA序列的宿主细胞DNA片段。研究结果表明,这种整合在E2与L2之间打断了HPV16 DNA链。并产生一个813碱基对的缺失,从而导致E6与E7的基因表达。在癌前损伤组织细胞就已整合有HPV16序列。Junyile等人[8](1988)研究发现从HPV16阳性的人的肿瘤组织细胞中获得的一段分子长度为4.4 kb的DNA片段(T4.4)(内含HPV16 E6启动子、E6E7、部分E1基因和宿主细胞DNA序列),具有完全转化NIH3T3细胞的活力,并已在转化的细胞内发现有大量的E6E7转录物存在。但是单独的HPV16片段或细胞DNA片段却无这种活性。当用多瘤病毒(polyomavirus)或SV40 DNA的多腺苷酸化序列(片段)取代T4.4中HPV16 DNA片段时,很少或者没有观察到转化活性的恢复。这些结果揭示含有从人肿瘤细胞基因组分离出来的HPV16E6E7 DNA片段具有转化潜力,这种潜力只有在HPV16 DNA被连接到宿主细胞DNA序列中才能实现。细胞DNA序列比以简单提供一个多腺苷酸化DNA片段更为复杂的方式发挥作用。
, http://www.100md.com
为了确定HPV16 DNA分子中那个基因在细胞转化中起作用,Masuo Yutsudo等人[9](1988)构建了含有HPV16早期开放阅读框架(EORF)不同亚基因片段的重组鼠源逆转录病毒的DNA,并测定了它们的恶性转化活性。结果发现E6和E7 ORF是HPV16的转化基因,前者在裸鼠体内决定致瘤性,而后者在软琼脂中影响细胞生长特性(例如细胞饱和密度与克隆形式);并发现在E1-E2 ORF区域可能存在一个抑癌基因,因为含有E1 ORF和E2 ORF片段加E6和E7 ORF片段的重组DNA的致瘤活性明显低于仅仅只有E6和E7 ORF片段DNA。国内张卫等人[10](1991)用HZIP16和HZIP16K 2种质粒,将HPV16的全早期区基因与E6、E7 ORF片段分别转入φ细胞,所产生的重组病毒能诱导NIH3T3细胞发生转化。获得的转化细胞具有接触抑制消失,非锚地依赖性生长,并可使裸鼠致瘤。结果表明,HPV16全EORF DNA片段具有诱导NIH3T3细胞恶性转化的活性。早期区基因中的E6-E7 ORFs也可单独诱导细胞恶性转化。为了探索HPV16长控制区(LCR)在E6E7 ORFs转化中的作用,候芷英等人[11]1994构建了含HPV16 E6E7 ORFs(nt83-855)片段和HPV16长控制区(LCR)加E6E7 ORFs片段(nt7007―7904/0-879)的逆转病毒载体pH21和pH18质粒,利用琼脂转染法,分别将它们导入病毒包装细胞pA317中,经过筛选获得G418抗性的病毒包装细胞,产生的重组病毒H21和H18感染的NIH3T3细胞都具有恶性细胞的形态学特征,并能使裸鼠形成肿瘤。Southern Blot杂交结果表明,上述两个基因片段都被整合到细胞基因组中。细胞转化结果说明,HPV16E6E7基因片段是HPV16转化NIH3T3细胞的关键早期基因,但其自身的LCR在细胞恶性转化过程中并没有显示出重要作用。盖其伟等人[12](1994)在体外用HPV16 DNA完整分子通过磷酸钙沉淀技术转录NIH3T3细胞,从而导致了NIH3T3细胞的恶性转化,并用Southern blot杂交证实HPV16 DNA分子以整合状态存在于NIH3T3细胞DNA中。
, 百拇医药
2.2 HPV16及其基因组与宿主细胞抑癌基因的关系 许多研究表明,HPV16及其基因组的致癌(肿瘤)性或细胞恶性转化作用是由于HPV16的有关基因通过影响宿主细胞抑癌基因表达产物生物活性来实现的。Dyson和Werness分别于1989年、1990年在《Science》杂志上发表论文[13,14],提出HPV16产生的两个病毒蛋白即E6、E7蛋白可能分别捆绑并抑制宿主细胞内的两个关键性的抗癌蛋白即P53蛋白和Rb蛋白。在正常细胞内,P53和Rb调控细胞周期与凋亡,P53与Rb表达产物的失活可导致宿主细胞永生化。E6蛋白可能与P53表达产物结合而导致P53降解失活,继之使含HPV16的宿主细胞生长失控。Magnus Non Knebel Doebejz等人[15](1994)用SW756宫颈癌细胞研究发现:E7蛋白与Rb结合可影响Rb与细胞E2F转录因子的结合,使E2F与Rb分离而具有转录激活功能,从而影响细胞的生长与分化,但在SW756衍生的细胞系中E7的情况并非如此。
, 百拇医药 P16(MTS1)肿瘤抑制基因是一个周期素依赖性激酶(cdk)的抑制基因,它能通过灭活cdk(cdk能使pRb磷酸化)使细胞周期减慢。Nakao-y等人[15]研究发现宫颈上皮细胞被HPV16永生化后,可观察到P16 RNA水平的急剧增加,而且,在随后的永生化过程中能观察到P16蛋白的表达。但是细胞恶性转化后,P16RNA与蛋白水平没有改变。这些结果表明,在恶性转化期间,不会发生P16-cdk-Rb串联体的失活,但在隐藏有HPV16的恶化前期(癌前期)损伤组织的永生化期间,则出现P16-cdk-Rb串联体的失活。
转化生长因子-β1(TGF-β1)是一个细胞分裂因子,对许多上皮细胞、内皮细胞、淋巴细胞等是一潜在性生长抑制剂,而对间质细胞与骨细胞则是促有丝分裂剂。Dey-A等人[17]研究评价了HPV16E6E7癌蛋白对TGF-β1启动子的作用。结果表明,HPV16E6明显诱导TGF-β1启动子活化(6倍于对照组),而HPV16E7则无明显作用。E6作用存在细胞型的特殊性,即仅仅在成纤维细胞中观察到它的作用,在上皮细胞则无。启动子序列分析表明,TGF-β1启动子的-109~-100序列中存在富含有GC的9个碱基对的片断即GGGGCGGGG,它是E6介导的反式活化的主要靶基因,HPV16E6蛋白的123~136位的3个氨基酸对TGF-β1启动子的反式活化至关重要。
, 百拇医药
E-Cadherin是位于染色体16q上,其编码蛋白有细胞表面粘附作用,也属于一种抑癌基因。HPV16使在培养基中的原发角质化细胞永生化,并抑制血清Ca刺激的分化。Wilding-J等人(1996)[18]用一个建立在HPV16基础上的角质化细胞永生化模型分析了ECad-herin的功能与表达的紊乱,也分析了正常角质化细胞所表达的Ca2+-依赖性细胞的细胞粘附分子及其相关蛋白。联合转染HPV16E6和E7所产生的永生化角质化细胞系能降低膜E-Cadherin表达,以及使α、β、γ细胞骨架蛋白(Catenin)从内膜到胞浆重新分布。上皮细胞生长因子受体(EGFr)的过度表达与被HPV16E6E7转染的角质化细胞中E-Cadherin/Catenin复合物的紊乱相关联,可能构成生长调节因子和吸附分子之间功能性相互影响的基础。
2.3 HPV16及其基因在宿主细胞DNA中的整合位点 脆性组氨酸三联体(FHIT)基因是一个被公认的肿瘤抑制基因,其中内含重叠的FRA3B(FRA3B是一个最有活性的染色体断裂点,位于染色体3p14.2处)。HPV16可以频繁地整合到FRA3B部位[19],并发现HPV16与FHIT/FRA3B区域中杂合性丢失有关。
, 百拇医药
Ets家族转录因子erg与ets-2表达变化与宫颈癌的发展有关。二者的表达变化与HPV16永生化的宫颈细胞和宫颈癌细胞系(CX16-2)染色体21q、22.2~22.3的基因改变相联系。而HPV16在CX16-2细胞中染色体21q 22.2~22.3易位断裂点发生整合[20]。
为了弄清楚在宫颈癌细胞中被HPV16整合所破坏的细胞染色体DNA位点的结构特性,Choo-KB等人[21](1996)用PCR技术直接扩增,分析了宫颈癌活检样品中细胞染色体DNA的4个位点。HPV16破坏的位点之一是2号染色体Map-2基因的位置,其他3个破坏位点分别是9号染色体PID,1号染色体PID-2与8号染色体PID-3,以上4对染色体携带着许多从前已测定过的HPV整合部位与染色体脆性部位和Myc癌基因。结果显示,HPV16基因组在宫颈癌细胞DNA中的整合位点多在染色体不稳定部位和具有转录活性的部位。
在许多宫颈肿瘤和从其衍生的细胞系中,HPV16 DNA序列能够整合到宿主细胞基因组中。Bauer-Hofmann-R等人[22](1996)用酶谱分析发现由于病毒DNA的整合大约有7.8 kb的宿主细胞DNA缺失。通过计算机分析,缺失的基因组区段2.3 kb的DNA序列和病毒整合位点上游的1.0 kb的序列与任何已知的人细胞DNA序列无明显同源性。这些发现提示,病毒整合发生在接近被公认的控制序列,病毒与宿主细胞DNA重组不会跟随有整合位点染色质结构上游的重组。RT-PCR实验表明,HPV的特殊转录跨越E2、E4、E5和位于HPV16控制区上游的L1/L2 ORFs。根据这些资料,综合起来考虑在SiHa细胞中,HPV16整合位点上游的宿主细胞DNA区域内含有影响位于HPV16上游控制区(URR)上游的HPV16 ORFs转录的控制元件。
, 百拇医药
3 抗HPV16癌基因的研究
为了评价核酶(Ribozyme)对CV-1细胞中E7基因表达的抑制作用,Huang-Y等人[23](1996)用带有1个降解E7 RNA的核酶和2个加工处理核酶的基因Rz523在5′和3′末端克隆到真核表达质粒PREP9中,在RSV-LTR启动的控制下,通过磷酸钙共沉淀法将其克隆产物质粒PRSV-Rz 523转染到CV-1细胞中。用点杂交法来检测G418抗性细胞中核酶的表达。在CV-1细胞中,稳定表达的核酶在9.0pmol/106细胞水平,其中有活性的核酶分子超过50fmol/106细胞。RNase保护性分析结果表明,在表达核酶CV-1细胞中的E7 RNA片段稳定含量显著地减少了90%。结果表明,用核酶抑制E7基因表达能使宫颈癌恶性表型逆转成为可能。
HPV16E6E7基因在宫颈癌致癌机制中起着中心作用,它们已被公认为与细胞中肿瘤抑制基因p53,p105与Rb等产物有相互作用,为了验证“E6E7在细胞恶性表型的维持中起主动作用”和“E6E7可能是抗基因治疗理论靶点”的假设,Madrigal-M等人[24](1997)用硫代寡脱氧核苷酸(phosphorothioate oligodeoxynucleotides)来攻打宫颈癌细胞系与原发肿瘤外殖体中HPV16E6E7,发现硫代寡脱氧核苷酸具有抗HPV16E6E7产生的增生作用。
, 百拇医药
在探索腺病毒反义RNA转录物对宫颈癌基因治疗可能性的研究中,Hamada-K等人[25](1996)将HPV16E6E7基因的反义RNA转录物通过重组腺病毒载体(Ad5 CMV-HPV16AS)导入隐含有HPV16的宫颈癌细胞中,分析了这些基因的表达对细胞生长与肿瘤生长的影响。Ad5 CMV-HPV16AS的基因框(盒)中含有巨细胞病毒启动子,反义HPV16E6E7基因和SV40多腺苷酸信号。使上述基因框插入到已修改了的腺病毒5的E1缺失区。结果表明,感染有Ad5 CMV-HPV16AS的细胞,其生长明显受到抑制。在体内研究中,给裸鼠注射Ad5 CMV-HPV16AS感染的SiHa细胞完全抑制了鼠的致瘤性。因而,以Ad5 CMV-HPV16AS形式转染含有HPV16E6E7反义RNA细胞,这可能是治疗HPV16阳性宫颈癌的一种具有潜力的新途径。
4 小结
综上所述,HPV16感染宿主细胞后,既可以HPV16全分子,也可以基因片段整合到宿主细胞DNA中(或染色体中),其表达蛋白可通过影响宿主细胞的一些抑癌(肿瘤)基因和相关细胞生长调控因子的生物活性来使被感染的宿主细胞恶性转化或发生癌变。目前普遍认为HPV16分子中E6E7基因是HPV16的致癌基因,并在裸鼠致瘤性实验中得到证实。通过抗HPV16癌基因的体外研究成果,由HPV16引起的宫颈癌的基因治疗存在广阔前景。
, 百拇医药
参考文献
[1] H.D.L.Birley.Human papillomaviruses,cervical cancer and the developing world.Annals of tropical medicine and parasitology,1995,89:453-463.
[2] 蔡红,姚,陈伟丽,等.多聚酶链反应检测宫颈癌HPV感染及分型的研究.中国病毒学,1994,9:291-296.
[3] 钟才高,Donald D.Anthony.HPV16及其基因诱导小鼠宫颈癌的毒理学研究.实用预防医学,1999,6:401-404.
[4] Lazo PA.The molecular genetics of cervical carcinoma.Br J cancer,1999,80:2008-2018.
, 百拇医药
[5] Christopher P Crum,Shannon Barber,James K Roche.Pathobiology of papillomavirusrelated cervical diseases:Prospects for immunodiagnosis.Clinical Microbiology Reviews,1991,4:270-285.
[6] Seedorf K,Kraemmer C,Duerst M,et al.Human papillomavirus type 16 DNA sequence.Virology,1985,145:181-185.
[7] Sylvie schneider-Maunoury,Odile croissant,Gerard Orth.Intergration of human papillomavirus type 16 DNA sequence:a possible early event in the progression of genital tumors.Journal of virology,1987,61:3295-3298.
, 百拇医药
[8] Jun-Yile,Vittorio Defendi.A viral-cellular junction fragment from a human papillomavirus type 16-positive tumour is competent in transformation of NIH3T3 cells.Journal of Virology,1988,62:4420-4426.
[9] Masuo Yutsudo,Yuji Okamoto,Akira Hakura.Functional dissociation of transforming genes of human papillomavirus type 16.Virology,1988,166:594-597.
[10] 张卫,司静懿,李昆,等.人乳头瘤病毒16型亚基因DNA体外转化功能的细胞学研究.病毒学报,1991,7:222-225.
, 百拇医药
[11] 候芷英,孙亚洲,商铭,等.人乳头瘤病毒16型E6E7 ORF转化作用的研究.中华实验和临床病毒学杂志,1994,8:112-115.
[12] 盖其伟,赫翠珍,姚庆英,等.人乳头瘤病毒16型DNA诱导NIH3T3细胞的恶性转化.首都医学院学报,1994,15:109-111.
[13] Dyson N,Howley PM,Munger K,et al.The human papillomavirus 16 E7 oncoprotein is able to bind the retinoblastoma gene product.Science,1989,243:934-936.
[14] Verness BA,Levine AJ,Howley PM.Association of human papillomavirus type 16 and 18 E6 proteins with p53.Science,1990,248:76-79.
, 百拇医药
[15] Magnus von knebel doeberitz,Claudia rittmuller,Frank Aengeneyndt,et al.Reversible repression of papillomavirus oncogene expression in cervical carcinoma cells:Consequences for the phenotype and E6-p53 and E7-pRb interactions.J Virol,1994,68:2811-2821.
[16] Nakao Y,Yang X,Yokoyama M,et al.Induction of p16 during immortalization by HPV16 and 18 and not during malignant transformation.Br J cancer,1997,75:1410-1416.
[17] Dey A,Atcha IA,Bagchi S.HPV16E6 oncoprotein stimulates the transforming growth factor-beta 1 promoter in fibroblasts through a specific GC-rich sequence.virology,1997,228:190-199.
, 百拇医药
[18] Wilding J,Vousden KH,Soutter WP,et al.E-cadherin transfection down-requlates the epidermal growth factor receptor and reverses the invasive phenotype of human papillomavirus-trasfected keratinocytes.Cancer Res,1996,56:5285-5292.
[19] Muller CY,O'Boyle Jd,Fong KM,et al.Abnormalities of fragile histidine triad genomic and complementary DNA in cervical cancer:association with human papillomavirus type. J Natl Cancer Inst,1998,90:433-439.
, 百拇医药
[20] Simpson S,Woodworth CD,Dipaolo JA.Altered expression of Erg and Ets-2 transcription factors is association with genetic changes at 21q22.2-22.3 in immortal and cervical carcinoma cell lines.Oncogene,1997,14(18):2149-2157.
[21] Choo KB,Chen CM,Han CP,et al.Molecular analysis of cellular loci disrupted by papillomavirus 16 intergration in cervical cancer:frequent viral intergration in topologically destabilized and transcriptionally active chromosomal regions.J Med Virol,1996,49:15-22.
, 百拇医药
[22] Bauer Hofmann R,Barghouts C,Auvinen E,et al.Genomic cloning and characterization of the nonoccupied allele correspounding to the intergration site of human papillomavirus type 16 DNA in the cervical cancer cell line SiHa.Virology,1996,217:33-41.
[23] Huang Y,Yong Y,Wang Y,et al.Stable expression of anti-HPV16E7-ribozyme in CV-1 Cell lines.Chin J Biotechnol,1996,12:215-220.
[24] Madrigal M,Janicak MF,Sevin BU,et al.In Vitro antigen therapy targeting HPV16E6 and E7 cervical carcinoma.Gynecol Oncol,1997,64:18-25.
[25] Hamada K,Sakaue M,Alemany R,et al.Adenovirus Mediated transfer of HPV16E6/E7 anti-sense RNA to human cervical cancer cells.Gynecol Oncol,1996,63:219-227.
(收稿日期:1999-10-18), http://www.100md.com