HPLC法测定连翘和银甲妇康液中连翘苷的含量
作者:王雪 王大果 晁若冰
单位:王大果(华西医科大学药学院 成都 610041 深圳市人民医院药剂科);晁若冰(华西医科大学药学院 成都 610041);王雪(华西医科大学药学院 成都 610041 成都军区总医院药学部 邮编 610083)
关键词:连翘苷;高效液相色谱法;银甲妇康液
药物分析杂志000206 摘要 目的:用反相高效液相色谱法测定连翘[Forsythia suspensa (Thunb.) Vahl.] 生药及银甲妇康液等含连翘制剂中连翘苷的含量。方法:样品用氧化铝柱净化后在ODS柱上分析,流动相为乙腈-水(22∶78),紫外检测波长为230 nm。结果:该方法的线性范围为0.15~1.95 μg,方法回收率为101.3%,RSD为1.3%。结论:该方法能消除中药制剂中复杂组分的干扰,准确地对连翘苷进行含量测定。
, http://www.100md.com
Determination of Phillyrin in Forsythia suspensa (Thunb.) and in Chinese Medicine Yinjia Fukang Solution by HPLC
Wang Xue, Wang Daguo and Chao Ruobing
(Department of Pharmacy, West China University of Medical Sciences, Chengdu 610041)
Abstract Objective:To determine the content of phillyrin in Crude Forsythia suspensa (Thumb.) and in Chinese Medicine Yinjia Fukang solution, Huanglian Shangqing pill, VC Yinqiao tablet by RP-HPLC.Method:Samples were purified by an Al2O3 column, then analyzed by RP-HPLC. The mobile phase was acetonitrile-water(22∶78). The UV detection wavelength was 230 nm.Result:A good linearity was obtained in the range of 0.15~1.95 μg (r=0.999 6), the average recovery was 101.3%(RSD=1.3%,n=15).Conclusion:This method can eliminate the interference of the complex components in the Chinese medicine, and results in an accuracy quantitative analysis of phillyrin.
, 百拇医药
Key words phillyrin, HPLC, Yinjia Fukang solution
银甲妇康液是由连翘、银花、鳖甲、草红藤等多味中药,经科学方法提取有效成分精制而成,是治疗盆腔炎、外阴道炎、尿路感染等疾病的中药口服制剂。其中的主药连翘具有清热止呕、清利肝胆、除湿退黄、通调三焦等多种作用[1]。连翘中的主要生物活性成分连翘苷(phillyrin)具有较强的抗菌作用,并能抑制CAMP磷酸二酯酶的活性[2],故以连翘苷作为指标成分,进行了含量测定。
以往的文献对连翘中连翘苷的测定方法有薄层色谱法[3]、梯度洗脱的RP-HPLC法[4]等。中药制剂中连翘苷的测定未曾报道过。我们用氧化铝柱层析对样品进行净化处理后,采用ODS柱、乙腈-水(22∶78)为流动相的RP-HPLC法,能够准确地对银甲妇康液中的连翘苷含量进行测定,阴性对照不干扰。我们还同时对连翘生药及其他含连翘的中药制剂黄连上清丸、维C银翘片中的连翘苷进行了测定,均取得较为满意的结果。
, http://www.100md.com
1 仪器与试药
日本岛津LC-6A高效液相色谱仪,SPD-6AV紫外检测器,C-R4A数据处理机。
连翘苷对照品(中国药品生物制品检定所),银甲妇康液(四川天府药业集团公司),维C银翘片(宜昌民康药业有限公司),黄连上清丸(成都军区总医院), 连翘(由四川天府药业集团公司命名,由成都军区总医院中医科孟海霞主管药师鉴定)。
乙腈(色谱用),重蒸馏水。
2 实验方法
2.1 供试品溶液的制备
2.1.1 银甲妇康液 精密量取银甲妇康液25 mL于水浴上挥至约10 mL,加甲醇10 mL,充分搅拌后过氧化铝柱(17 cm×2 cm,中性氧化铝20 g),用50%的甲醇35 mL少量多次洗脱,收集洗脱液于水浴上挥干,残渣用50%的甲醇溶解后转移至25 mL容量瓶中,加50%甲醇稀释至刻度,过滤,弃去初滤液,收集续滤液备用。
, 百拇医药
2.1.2 银甲妇康液阴性对照 去掉处方中的连翘生药后,照生产工艺制备得到银甲妇康空白溶液(由天府药业集团有限公司提供),然后按“2.1.1”项的方法制备得到阴性对照液。
2.1.3 连翘 取适量连翘生药粉碎后,精密称取生药粉末约0.5 g,加甲醇15 mL超声提取30 min后,转移至25 mL容量瓶中,加甲醇稀释至刻度,摇匀后过滤,弃去初滤液,取续滤液备用。
2.1.4 黄连上清丸 精密称取黄连上清丸细粉约6.0 g,精密加入甲醇25 mL,超声提取30 min,过滤,精密量取续滤液15 mL过氧化铝柱(17 cm×2 cm,中性氧化铝20 g),用50%的甲醇35 mL少量多次洗脱,收集洗脱液,水浴挥干后,精密加入50%的甲醇10 mL溶解,过滤,取续滤液即得。
2.1.5 维C银翘片 取维C银翘片20片,去糖衣后,精密称定。研细,精密称取片粉适量(约相当于维C银翘片5片),精密加入甲醇25 mL,超声提取30 min,以下操作同“2.1.4”项。
, http://www.100md.com
2.2 连翘苷对照品溶液的配制 精密称取连翘苷对照品约25 mg,置50 mL容量瓶中,加甲醇溶解并稀释至刻度,摇匀,即得浓度约为0.50 mg。mL-1的对照品贮备液。精密量取对照品贮备液15 mL于50 mL容量瓶中,用50%的甲醇稀释到刻度,摇匀,即得浓度约为0.15mg。mL-1的对照品溶液。
2.3 色谱条件 经试验后选定的色谱条件为色谱柱:Shim-Pack CLC ODS柱(150 mm×6.0 mm),柱温:30 ℃;流动相:乙腈-水(22∶78),流速:1.0 mL。min-1;检测波长为230 nm。
3 实验结果
3.1 色谱图 在选定条件下, 连翘苷对照品、银甲妇康液、连翘以及银甲妇康液连翘阴性对照的色谱图见图1。可见, 连翘苷出峰处阴性对照无干扰。
, http://www.100md.com
图1 连翘苷和银甲妇康液的色谱图
a.连翘苷对照品 b.连翘苷阴性对照
c.银甲妇康液 d.连翘药材
1.连翘苷(23.06 min)
3.2 线性与范围 分别取连翘苷对照品溶液1,3,5,7,9,11,13 μL进样,样品平行操作,记录色谱图,以峰面积对连翘苷的进样量X(μg)作线性回归,得到线性方程为:
A=8.446+759.6 X r=0.999 6
说明连翘苷进样量在0.15~1.95 μg范围内线性较好。
3.3 回收试验
, 百拇医药 3.3.1 净化回收率 分别精密量取连翘苷对照品溶液2 mL,分别加水2 mL,照银甲妇康液样品预处理方法制备试验溶液。另精密量取同一对照品贮备液2 mL,置25 mL容量瓶中,用50%的甲醇稀释到刻度,即得对照溶液。供试溶液和对照溶液分别进样10 μL,记录色谱图,根据所得峰面积,计算净化回收率。平均净化回收率为99.1%(n=6),RSD=0.74%。说明采用选定方法净化样品有很好的净化回收率。
3.3.2 加样回收率 取银甲妇康空白溶液(银甲妇康液的处方中去掉连翘,但制备工艺相同)25 mL,分别准确加入一定量的连翘苷对照品,照银甲妇康液样品预处理方法制备得试验溶液。进样10 μL,另取连翘苷对照溶液1,3,5,7,9,11,13 μL进样,按标准曲线法测定并计算加样回收率,结果见表1。平均回收率为101.3%,RSD=1.3%,n=15。
表1 回收率测定结果(n=3) 加入量/mg
, 百拇医药 回收率/%
RSD/%
0.492
102.3
1.15
0.98
100.6
1.62
1.48
100.8
1.25
1.97
100.3
, 百拇医药
0.76
2.46
102.3
0.23
3.4 重复性试验 分别精密量取同一供试品溶液25 mL,照预处理项下供试品溶液制备方法制备试验溶液,取10 μL进样,测得平均峰面积值为A=552 180,RSD为0.54%(n=6)。
3.5 稳定性试验 于0,2,4,6,8,12 h取同一供试品溶液和对照品溶液各10 μL,分别进样2次,计算日内RSD分别为1.11%和0.91%。再于第1,2,3,4 d分别取同一供试品溶液和对照品溶液各10 μL,进样2次,计算日间RSD分别为2.54%和0.84%。
3.6 进样精密度试验 取对照品溶液,重复进样6次,根据峰面积值,计算得平均峰面积A=737 880,RSD为0.85%。
, 百拇医药
3.7 系统适用性试验 照“2.1.1”项下方法制备样品溶液,照“2.3”项下的色谱条件进样1次,以连翘苷峰计算,测得柱效为9.2×103,连翘苷与其前后杂质的分离度分别为8.85和5.31。
3.8 样品测定 采用前述的样品预处理方法和色谱条件,分别测定了5个批号的银甲妇康液中连翘苷的含量以及连翘生药中连翘苷含量,结果见表2。表2 不同样品中连翘苷的含量(n=3) 样品
平均含量/mg。mL-1
RSD/%
银甲妇康液970901
0.584
1.11
, http://www.100md.com
970904
0.512
0.15
971001
0.757
1.19
971002
0.892
0.42
950903
0.255
0.61
连翘
, 百拇医药
0.0120(mg。mg-1)
1.35
4 讨论
4.1 我们曾经采用过甲醇(含1%的四氢呋喃)-水(含0.01 mol。L-1磷酸二氢钠,pH 3.2)(40∶60)的系统,不同比例的乙腈-甲醇-水系统,以及不同比例的乙腈-水系统,结果表明:乙腈-水为22∶78时, 连翘苷峰形对称,分离完全,阴性对照不干扰。我们同时还发现,乙腈比例微小的变化会较显著地影响连翘苷的保留时间。
4.2 我们还尝试采用选定的方法对连翘苷的其它2种制剂:黄连上清丸、维C银翘片中的连翘苷进行了分离、测定。色谱图见图2,测定结果见表3。可知, 连翘苷的峰形对称,与其他峰的分离较好,达到基线分离。说明实验方法可用于此类制剂中的连翘苷含量测定。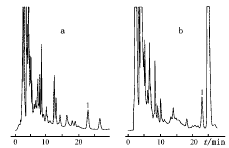
, 百拇医药
图2 连翘制剂的色谱图
a.黄连上清丸 b.维C银翘片
1.连翘苷(23.15 min)
表3 其它连翘制剂中连翘苷的测定结果(n=3) 样品
平均含量
RSD/%
黄连上清丸
0.42 mg。g-1
1.35
维C银翘片
0.58 mg每片
, 百拇医药
1.26
4.3 在样品预处理方法中,我们曾尝试采用过液液萃取法和硅胶柱作为层析柱的方法。前者由于净化不完全,引入杂质多而放弃。后者则会使连翘苷在柱上的保留性较强,不易被完全洗脱。采用中性氧化铝柱,连翘苷能很容易地被50%甲醇洗脱,而中药制剂中黄酮类等干扰组分能够有效地被保留在柱上,用于以上几个品种的预处理,净化效果好,故确定采用氧化铝柱。
4.4 在对不同批号的银甲妇康液中连翘苷测定时,发现其含量差异较大。
参考文献
1,丁冈,刘延泽.中药连翘甙及其同属植物的研究近况.中药材,1994,17(10):42
2,刘东雷,李会轻,徐绥绪. 连翘属植物化学成分的研究进展.沈阳药科大学学报,1995,12(4):301
3,梁文藻,董慧,涂国士. 连翘成分分析Ⅰ.连翘甙和连翘脂素的分离鉴定和测定.药物分析杂志,1985,5(1):1
4,崔燕岩,冯少勇,赵光等. 连翘有效成分的HPLC法测定.药学学报,1992,27(8):603
(本文于1999年7月26日修改回), 百拇医药
单位:王大果(华西医科大学药学院 成都 610041 深圳市人民医院药剂科);晁若冰(华西医科大学药学院 成都 610041);王雪(华西医科大学药学院 成都 610041 成都军区总医院药学部 邮编 610083)
关键词:连翘苷;高效液相色谱法;银甲妇康液
药物分析杂志000206 摘要 目的:用反相高效液相色谱法测定连翘[Forsythia suspensa (Thunb.) Vahl.] 生药及银甲妇康液等含连翘制剂中连翘苷的含量。方法:样品用氧化铝柱净化后在ODS柱上分析,流动相为乙腈-水(22∶78),紫外检测波长为230 nm。结果:该方法的线性范围为0.15~1.95 μg,方法回收率为101.3%,RSD为1.3%。结论:该方法能消除中药制剂中复杂组分的干扰,准确地对连翘苷进行含量测定。
, http://www.100md.com
Determination of Phillyrin in Forsythia suspensa (Thunb.) and in Chinese Medicine Yinjia Fukang Solution by HPLC
Wang Xue, Wang Daguo and Chao Ruobing
(Department of Pharmacy, West China University of Medical Sciences, Chengdu 610041)
Abstract Objective:To determine the content of phillyrin in Crude Forsythia suspensa (Thumb.) and in Chinese Medicine Yinjia Fukang solution, Huanglian Shangqing pill, VC Yinqiao tablet by RP-HPLC.Method:Samples were purified by an Al2O3 column, then analyzed by RP-HPLC. The mobile phase was acetonitrile-water(22∶78). The UV detection wavelength was 230 nm.Result:A good linearity was obtained in the range of 0.15~1.95 μg (r=0.999 6), the average recovery was 101.3%(RSD=1.3%,n=15).Conclusion:This method can eliminate the interference of the complex components in the Chinese medicine, and results in an accuracy quantitative analysis of phillyrin.
, 百拇医药
Key words phillyrin, HPLC, Yinjia Fukang solution
银甲妇康液是由连翘、银花、鳖甲、草红藤等多味中药,经科学方法提取有效成分精制而成,是治疗盆腔炎、外阴道炎、尿路感染等疾病的中药口服制剂。其中的主药连翘具有清热止呕、清利肝胆、除湿退黄、通调三焦等多种作用[1]。连翘中的主要生物活性成分连翘苷(phillyrin)具有较强的抗菌作用,并能抑制CAMP磷酸二酯酶的活性[2],故以连翘苷作为指标成分,进行了含量测定。
以往的文献对连翘中连翘苷的测定方法有薄层色谱法[3]、梯度洗脱的RP-HPLC法[4]等。中药制剂中连翘苷的测定未曾报道过。我们用氧化铝柱层析对样品进行净化处理后,采用ODS柱、乙腈-水(22∶78)为流动相的RP-HPLC法,能够准确地对银甲妇康液中的连翘苷含量进行测定,阴性对照不干扰。我们还同时对连翘生药及其他含连翘的中药制剂黄连上清丸、维C银翘片中的连翘苷进行了测定,均取得较为满意的结果。
, http://www.100md.com
1 仪器与试药
日本岛津LC-6A高效液相色谱仪,SPD-6AV紫外检测器,C-R4A数据处理机。
连翘苷对照品(中国药品生物制品检定所),银甲妇康液(四川天府药业集团公司),维C银翘片(宜昌民康药业有限公司),黄连上清丸(成都军区总医院), 连翘(由四川天府药业集团公司命名,由成都军区总医院中医科孟海霞主管药师鉴定)。
乙腈(色谱用),重蒸馏水。
2 实验方法
2.1 供试品溶液的制备
2.1.1 银甲妇康液 精密量取银甲妇康液25 mL于水浴上挥至约10 mL,加甲醇10 mL,充分搅拌后过氧化铝柱(17 cm×2 cm,中性氧化铝20 g),用50%的甲醇35 mL少量多次洗脱,收集洗脱液于水浴上挥干,残渣用50%的甲醇溶解后转移至25 mL容量瓶中,加50%甲醇稀释至刻度,过滤,弃去初滤液,收集续滤液备用。
, 百拇医药
2.1.2 银甲妇康液阴性对照 去掉处方中的连翘生药后,照生产工艺制备得到银甲妇康空白溶液(由天府药业集团有限公司提供),然后按“2.1.1”项的方法制备得到阴性对照液。
2.1.3 连翘 取适量连翘生药粉碎后,精密称取生药粉末约0.5 g,加甲醇15 mL超声提取30 min后,转移至25 mL容量瓶中,加甲醇稀释至刻度,摇匀后过滤,弃去初滤液,取续滤液备用。
2.1.4 黄连上清丸 精密称取黄连上清丸细粉约6.0 g,精密加入甲醇25 mL,超声提取30 min,过滤,精密量取续滤液15 mL过氧化铝柱(17 cm×2 cm,中性氧化铝20 g),用50%的甲醇35 mL少量多次洗脱,收集洗脱液,水浴挥干后,精密加入50%的甲醇10 mL溶解,过滤,取续滤液即得。
2.1.5 维C银翘片 取维C银翘片20片,去糖衣后,精密称定。研细,精密称取片粉适量(约相当于维C银翘片5片),精密加入甲醇25 mL,超声提取30 min,以下操作同“2.1.4”项。
, http://www.100md.com
2.2 连翘苷对照品溶液的配制 精密称取连翘苷对照品约25 mg,置50 mL容量瓶中,加甲醇溶解并稀释至刻度,摇匀,即得浓度约为0.50 mg。mL-1的对照品贮备液。精密量取对照品贮备液15 mL于50 mL容量瓶中,用50%的甲醇稀释到刻度,摇匀,即得浓度约为0.15mg。mL-1的对照品溶液。
2.3 色谱条件 经试验后选定的色谱条件为色谱柱:Shim-Pack CLC ODS柱(150 mm×6.0 mm),柱温:30 ℃;流动相:乙腈-水(22∶78),流速:1.0 mL。min-1;检测波长为230 nm。
3 实验结果
3.1 色谱图 在选定条件下, 连翘苷对照品、银甲妇康液、连翘以及银甲妇康液连翘阴性对照的色谱图见图1。可见, 连翘苷出峰处阴性对照无干扰。
, http://www.100md.com
图1 连翘苷和银甲妇康液的色谱图
a.连翘苷对照品 b.连翘苷阴性对照
c.银甲妇康液 d.连翘药材
1.连翘苷(23.06 min)
3.2 线性与范围 分别取连翘苷对照品溶液1,3,5,7,9,11,13 μL进样,样品平行操作,记录色谱图,以峰面积对连翘苷的进样量X(μg)作线性回归,得到线性方程为:
A=8.446+759.6 X r=0.999 6
说明连翘苷进样量在0.15~1.95 μg范围内线性较好。
3.3 回收试验
, 百拇医药 3.3.1 净化回收率 分别精密量取连翘苷对照品溶液2 mL,分别加水2 mL,照银甲妇康液样品预处理方法制备试验溶液。另精密量取同一对照品贮备液2 mL,置25 mL容量瓶中,用50%的甲醇稀释到刻度,即得对照溶液。供试溶液和对照溶液分别进样10 μL,记录色谱图,根据所得峰面积,计算净化回收率。平均净化回收率为99.1%(n=6),RSD=0.74%。说明采用选定方法净化样品有很好的净化回收率。
3.3.2 加样回收率 取银甲妇康空白溶液(银甲妇康液的处方中去掉连翘,但制备工艺相同)25 mL,分别准确加入一定量的连翘苷对照品,照银甲妇康液样品预处理方法制备得试验溶液。进样10 μL,另取连翘苷对照溶液1,3,5,7,9,11,13 μL进样,按标准曲线法测定并计算加样回收率,结果见表1。平均回收率为101.3%,RSD=1.3%,n=15。
表1 回收率测定结果(n=3) 加入量/mg
, 百拇医药 回收率/%
RSD/%
0.492
102.3
1.15
0.98
100.6
1.62
1.48
100.8
1.25
1.97
100.3
, 百拇医药
0.76
2.46
102.3
0.23
3.4 重复性试验 分别精密量取同一供试品溶液25 mL,照预处理项下供试品溶液制备方法制备试验溶液,取10 μL进样,测得平均峰面积值为A=552 180,RSD为0.54%(n=6)。
3.5 稳定性试验 于0,2,4,6,8,12 h取同一供试品溶液和对照品溶液各10 μL,分别进样2次,计算日内RSD分别为1.11%和0.91%。再于第1,2,3,4 d分别取同一供试品溶液和对照品溶液各10 μL,进样2次,计算日间RSD分别为2.54%和0.84%。
3.6 进样精密度试验 取对照品溶液,重复进样6次,根据峰面积值,计算得平均峰面积A=737 880,RSD为0.85%。
, 百拇医药
3.7 系统适用性试验 照“2.1.1”项下方法制备样品溶液,照“2.3”项下的色谱条件进样1次,以连翘苷峰计算,测得柱效为9.2×103,连翘苷与其前后杂质的分离度分别为8.85和5.31。
3.8 样品测定 采用前述的样品预处理方法和色谱条件,分别测定了5个批号的银甲妇康液中连翘苷的含量以及连翘生药中连翘苷含量,结果见表2。表2 不同样品中连翘苷的含量(n=3) 样品
平均含量/mg。mL-1
RSD/%
银甲妇康液970901
0.584
1.11
, http://www.100md.com
970904
0.512
0.15
971001
0.757
1.19
971002
0.892
0.42
950903
0.255
0.61
连翘
, 百拇医药
0.0120(mg。mg-1)
1.35
4 讨论
4.1 我们曾经采用过甲醇(含1%的四氢呋喃)-水(含0.01 mol。L-1磷酸二氢钠,pH 3.2)(40∶60)的系统,不同比例的乙腈-甲醇-水系统,以及不同比例的乙腈-水系统,结果表明:乙腈-水为22∶78时, 连翘苷峰形对称,分离完全,阴性对照不干扰。我们同时还发现,乙腈比例微小的变化会较显著地影响连翘苷的保留时间。
4.2 我们还尝试采用选定的方法对连翘苷的其它2种制剂:黄连上清丸、维C银翘片中的连翘苷进行了分离、测定。色谱图见图2,测定结果见表3。可知, 连翘苷的峰形对称,与其他峰的分离较好,达到基线分离。说明实验方法可用于此类制剂中的连翘苷含量测定。
, 百拇医药
图2 连翘制剂的色谱图
a.黄连上清丸 b.维C银翘片
1.连翘苷(23.15 min)
表3 其它连翘制剂中连翘苷的测定结果(n=3) 样品
平均含量
RSD/%
黄连上清丸
0.42 mg。g-1
1.35
维C银翘片
0.58 mg每片
, 百拇医药
1.26
4.3 在样品预处理方法中,我们曾尝试采用过液液萃取法和硅胶柱作为层析柱的方法。前者由于净化不完全,引入杂质多而放弃。后者则会使连翘苷在柱上的保留性较强,不易被完全洗脱。采用中性氧化铝柱,连翘苷能很容易地被50%甲醇洗脱,而中药制剂中黄酮类等干扰组分能够有效地被保留在柱上,用于以上几个品种的预处理,净化效果好,故确定采用氧化铝柱。
4.4 在对不同批号的银甲妇康液中连翘苷测定时,发现其含量差异较大。
参考文献
1,丁冈,刘延泽.中药连翘甙及其同属植物的研究近况.中药材,1994,17(10):42
2,刘东雷,李会轻,徐绥绪. 连翘属植物化学成分的研究进展.沈阳药科大学学报,1995,12(4):301
3,梁文藻,董慧,涂国士. 连翘成分分析Ⅰ.连翘甙和连翘脂素的分离鉴定和测定.药物分析杂志,1985,5(1):1
4,崔燕岩,冯少勇,赵光等. 连翘有效成分的HPLC法测定.药学学报,1992,27(8):603
(本文于1999年7月26日修改回), 百拇医药