Alport 综合征一例
作者:罗岩 张承芬
单位:(罗岩Email: luoyan @ bigfoot.com)(100730 中国医学科学院 中国协和医科大学;北京协和医院眼科)
关键词:
中华眼科杂志000227 患者男,21岁。因双眼视力逐渐下降7年,蛋白尿、听力下降4年,高血压1年,于1998年5月入我院肾病内科诊治。既往无眼部疾病史。母亲曾有“肾盂肾炎”病史,已治愈。其他家族病史不详。体检:血压160/110 mm Hg(1 mm Hg=0.133 kPa);轻度Cushing征外貌,余无异常。眼部检查:裸眼视力右眼0.2,左眼0.4;矫正视力右眼0.7(-11.0 DS +0.75 DC×90°),左眼0.7(-10.0 DS+0.75 DC×90°);双眼晶状体前表面中央区局限性前突,呈锥形,囊膜下可见小斑点状白色混浊(图1,2);双眼视盘色泽正常,边界清晰,视网膜血管大致正常,后极部黄斑区周围可见散在的黄白色点状颗粒,黄斑中心窝反光可见(图3,4);眼底血管荧光造影检查示双眼黄斑拱环结构破坏。辅助检查:(1)耳科电测听:双耳对称性感音神经性耳聋(中、重度);(2)肾活检报告:光镜示系膜增殖性肾小球肾炎改变(重度);免疫荧光检查为阴性;电镜示肾小球基底膜广泛变厚、劈裂;(3)眼电图、视网膜电图未见异常表现;(4)尿蛋白50 mg/L,血尿素氮 7.2 mmol/L, 血清肌酐185.6 μmol/L。诊断:Alport综合征。
+0.75 DC×90°),左眼0.7(-10.0 DS+0.75 DC×90°);双眼晶状体前表面中央区局限性前突,呈锥形,囊膜下可见小斑点状白色混浊(图1,2);双眼视盘色泽正常,边界清晰,视网膜血管大致正常,后极部黄斑区周围可见散在的黄白色点状颗粒,黄斑中心窝反光可见(图3,4);眼底血管荧光造影检查示双眼黄斑拱环结构破坏。辅助检查:(1)耳科电测听:双耳对称性感音神经性耳聋(中、重度);(2)肾活检报告:光镜示系膜增殖性肾小球肾炎改变(重度);免疫荧光检查为阴性;电镜示肾小球基底膜广泛变厚、劈裂;(3)眼电图、视网膜电图未见异常表现;(4)尿蛋白50 mg/L,血尿素氮 7.2 mmol/L, 血清肌酐185.6 μmol/L。诊断:Alport综合征。
讨论 Alport综合征又名眼耳肾综合征;遗传性血尿肾病耳聋综合征。诊断依据:(1)阳性家族史,遗传方式包括X染色体显性遗传(最多)、常染色体显性遗传(次之)、常染色体隐性遗传(少见);(2)肾脏病变(血尿、进行性肾功能不全);(3)双耳病变(高频性感音神经性耳聋);(4)双眼对称性病变(前锥形晶状体并发高度近视;囊膜下白内障;点状视网膜病变,即黄斑区周围视网膜有明亮的白色或黄色致密微粒);(5)肾脏活检及病理检查时电镜下所见肾小球基底膜(GBM)异常为本病确诊依据;免疫荧光检查通常为阴性(因为无体液免疫反应参与致病)可作为鉴别诊断依据;光镜检查无特异性。神经、肌肉、血液、内分泌系统都可发生病变,亦不具特异性。 该病发病机制目前考虑为基因异常导致IV型胶原变性,从而引起全身性基底膜病变。此病为基因异常所致,无特殊治疗措施。男性患者更易发生耳聋和肾功能衰竭,预后较差。
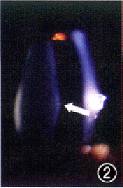

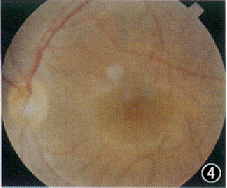
图1 右眼晶状体前表面中央区局限性前突,呈锥形,囊膜下可见小斑点状白色混浊(箭头) 图2 左眼晶状体前表面特征同图1 图3右眼视盘色泽正常,边界清晰,视网膜血管大致正常,后极部黄斑区周围可见散在的黄白色点状颗粒,黄斑中心凹反光可见 图4 左眼眼底特征同图3
(收稿日期:1999-06-24), http://www.100md.com
单位:(罗岩Email: luoyan @ bigfoot.com)(100730 中国医学科学院 中国协和医科大学;北京协和医院眼科)
关键词:
中华眼科杂志000227 患者男,21岁。因双眼视力逐渐下降7年,蛋白尿、听力下降4年,高血压1年,于1998年5月入我院肾病内科诊治。既往无眼部疾病史。母亲曾有“肾盂肾炎”病史,已治愈。其他家族病史不详。体检:血压160/110 mm Hg(1 mm Hg=0.133 kPa);轻度Cushing征外貌,余无异常。眼部检查:裸眼视力右眼0.2,左眼0.4;矫正视力右眼0.7(-11.0 DS
+0.75 DC×90°),左眼0.7(-10.0 DS+0.75 DC×90°);双眼晶状体前表面中央区局限性前突,呈锥形,囊膜下可见小斑点状白色混浊(图1,2);双眼视盘色泽正常,边界清晰,视网膜血管大致正常,后极部黄斑区周围可见散在的黄白色点状颗粒,黄斑中心窝反光可见(图3,4);眼底血管荧光造影检查示双眼黄斑拱环结构破坏。辅助检查:(1)耳科电测听:双耳对称性感音神经性耳聋(中、重度);(2)肾活检报告:光镜示系膜增殖性肾小球肾炎改变(重度);免疫荧光检查为阴性;电镜示肾小球基底膜广泛变厚、劈裂;(3)眼电图、视网膜电图未见异常表现;(4)尿蛋白50 mg/L,血尿素氮 7.2 mmol/L, 血清肌酐185.6 μmol/L。诊断:Alport综合征。讨论 Alport综合征又名眼耳肾综合征;遗传性血尿肾病耳聋综合征。诊断依据:(1)阳性家族史,遗传方式包括X染色体显性遗传(最多)、常染色体显性遗传(次之)、常染色体隐性遗传(少见);(2)肾脏病变(血尿、进行性肾功能不全);(3)双耳病变(高频性感音神经性耳聋);(4)双眼对称性病变(前锥形晶状体并发高度近视;囊膜下白内障;点状视网膜病变,即黄斑区周围视网膜有明亮的白色或黄色致密微粒);(5)肾脏活检及病理检查时电镜下所见肾小球基底膜(GBM)异常为本病确诊依据;免疫荧光检查通常为阴性(因为无体液免疫反应参与致病)可作为鉴别诊断依据;光镜检查无特异性。神经、肌肉、血液、内分泌系统都可发生病变,亦不具特异性。 该病发病机制目前考虑为基因异常导致IV型胶原变性,从而引起全身性基底膜病变。此病为基因异常所致,无特殊治疗措施。男性患者更易发生耳聋和肾功能衰竭,预后较差。
图1 右眼晶状体前表面中央区局限性前突,呈锥形,囊膜下可见小斑点状白色混浊(箭头) 图2 左眼晶状体前表面特征同图1 图3右眼视盘色泽正常,边界清晰,视网膜血管大致正常,后极部黄斑区周围可见散在的黄白色点状颗粒,黄斑中心凹反光可见 图4 左眼眼底特征同图3
(收稿日期:1999-06-24), http://www.100md.com