呋塞米对α1肾上腺素受体亚型引起的Ca2+内流的影响
李劲梁 关永源 韩启德 贺华 丘钦英
摘 要 目的 探讨Cl-通道开放在不同α1肾上腺素受体亚型引起的Ca2+内流中的作用。方法 采用Fura-2/AM荧光分光光度法,测定胞浆游离Ca2+浓度,观察Cl-通道阻断剂呋塞米,钙通道阻断剂SK&F96365和硝苯地平对不同α1肾上腺素受体亚型(α1A、α1B-AR)引起的Ca2+内流的影响,并加以比较。结果 2.5,5和10 μmol.L-1呋塞米呈浓度依赖性地抑制了α1A和α1B-AR亚型引起的Ca2+内流;对α1A-AR引起的Ca2+内流的抑制率分别为8.3%±6.1%,23.8%±14.8%和40.7%±19.3%,对α1B-AR引起的Ca2+内流的抑制率分别为12.0%±5.4%,19.2%±4.9%和31.9%±4.9%。α1A-AR引起的Ca2+内流被最大抑制浓度(20 μmol.L-1)的SK&F96365抑制后,不能被10 μmol.L-1呋塞米进一步抑制;而α1B-AR引起的Ca2+内流被20 μmol.L-1 SK&F96365抑制后,仍能被10 μmol.L-1呋塞米进一步抑制,抑制率为6.6%±3.2%。结论 参与α1A和α1B-AR引起的Ca2+内流的Cl-通道是非同一性的。
, 百拇医药
关键词:α1肾上腺素受体 Ca2+内流 Cl-通道 呋塞米 SK&F96365 硝苯地平
细胞内游离Ca2+水平的升高是许多生理、病理过程的起始信号,象血管的收缩,细胞的分泌,以及某些疾病如高血压、动脉粥样硬化等的发生都与之密切相关[1]。而受体介导的持续性Ca2+内流(re-ceptor-operated-Ca2+-influx;ROC)则是引起胞浆Ca2+水平升高的重要形式[2]。受体激动后,通过活化磷酯酰肌醇系统,水解出IP3,引起细胞内Ca2+池的Ca2+释放,进而触发因Ca2+释放引起的Ca2+内流(Ca2+-release-activated-Ca2+-influx; CRAC)。但是,Ca2+释放是通过什么机制引起Ca2+内流的,目前尚不清楚。Cl-离子是细胞外液中的主要阴离子。近来的研究表明,Cl-通道的开放对于CRAC的产生具有重要的意义[3~5]。在外周血管平滑肌的实验表明,Cl-通道参与了α1肾上腺素受体(α1-AR)激活引起的Ca2+内流[6]。神经递质去甲肾上腺素(noradrenaline;NA)激动α1-AR后,引起Ca2+释放,胞浆Ca2+水平升高,导致Cl-通道的开放,后者能进一步引起胞膜Ca2+通道的开放,产生持续性的Ca2+内流。选择性的α1-AR阻断剂哌唑嗪和一些Cl-通道阻断剂(如DIDS,呋塞米等)能不同程度地抑制Ca2+释放激活的Cl-通道开放[6]。本实验室的研究已经证实,参与ROC和电压依赖性的Ca2+内流(voltage-depedent- Ca2+-influx;VDC)的Cl-通道是非同一性的(未发表)。不同的α1-AR亚型引起的Ca2+内流中,参与的Cl-通道是否也存在这种情况,还有待进一步研究。
, 百拇医药
本研究以转染了α1A、α1B-AR cDNA的HEK293细胞为模型,采用Fura-2/AM荧光探针,测定胞浆游离Ca2+浓度([Ca2+]i),观察Cl- 通道阻断剂呋塞米及ROC通道阻断剂SK&F96365和VDC通道阻断剂硝苯地平对α1A、α1B-AR引起的Ca2+内流的影响,并加以比较。
1 材料与方法
1.1 实验材料 用牛α1A-AR和仓鼠α1B-AR cDNA转染的HEK293细胞(分别定义为HEKα1A和HEKα1B细胞)均由Dr. Minneman馈赠。Fura-2/AM,牛血清白蛋白(BSA),histidinol, hygromycin购于Borhringer Mannheim公司。肾上腺素(adrenaline;Adr),呋塞米(furosemide),硝苯地平(nifedipine), triton X-100, EGTA, HEPES购于Sigma公司。SK&F96365 购于Biomol 公司。DMEM/F12购于Gibico公司。小牛血清由本校微生物教研室提供。其余试剂均为国产分析纯。
, http://www.100md.com
1.2 实验仪器 RF-5 000荧光分光光度计(日本岛津公司),CO2培养箱(美国Precision公司),倒置显微镜(重庆光学仪器厂)。
1.3 实验方法
1.3.1 细胞培养 HEK293细胞用含10%(V/V)的小牛血清,100×103 U.L-1青霉素和125 mg.L-1链霉素的DMLE M/F12培养基,置于5% CO2,37℃的细胞培养箱内培养。HEKα1A和HEK1B细 胞分别用含有0.60 g.L-1 histidinol和0.05 g.L-1 hygromycin筛选抗生 素的培养基(含10%小牛血清的DMEM/F12)在上述条件下培养。细胞传代时,先用PBS(mmol.L -1: NaCl 130.0, KCl 2.5,Na2HPO4 10.0, KH2PO4 1.5,pH 7.4)洗1 次,加消化液(0.05% trypsin和0.02% EDTA溶于PBS,pH 7.4)消化30 s,吸弃消化液, 加入DMEM/F12终止消化,吹打后接种传代。
, 百拇医药
1.3.2 细胞[Ca2+]i测定 按本实验室已建立的方法进行[7],简述如下:当细胞生长至70%~ 90%融合时收获。用消化液消化细胞后,加入DMEM/F12(含0.05% BSA)终止消化。离心后 弃去上清,用含1 μmol.L-1 Fura-2/AM的DMEM/F12 (含0.05% BSA)重悬细胞,置 37℃、5% CO2条件下温育30 min。离心洗去细胞外的Fura-2/AM ,用BSS缓冲液(mmol.L -1: NaCl 130.0,KCl 5.0,MgCl2 1.0,CaCl2 1.5,HEPES 20.0, Glucose 10 .0, BSA 1.0 g, pH 7.4)重悬细胞,调整细胞密度至3×106个细胞/ml,在37℃恒温、磁力搅拌下进行荧光测定细胞[Ca2+]i。测定所用激发光波长为340 nm和380 nm,发射光波长为510 nm;测定结果由计算机自动记录,计算,并打印。所有实验试剂在有效剂量范围内都经证明无荧光干扰。
, 百拇医药
1.4 统计方法 实验结果以±s表示;组间及给药前后的[Ca2+]i差异用组间或配对t检验;P<0.05认为在统计学上差异有显著性。Adr引起的Ca2+净释放量和Ca2+净内流量及药物对Ca2+内流的抑制率按下列公式计算:
Ca2+净释放量([Ca2+] of release)=快速上升相[Ca2+]i-静息[Ca2+]i
Ca2+净内流量([Ca2+] of influx)=平台相[Ca2+]i-静息[Ca2+]i
, http://www.100md.com 抑制率(%)=(加药前平台相[Ca2+]i-加药后平台相[Ca2+]i)/(加药前平台相[Ca2+]i-静息[Ca2+]i)×100%
2 结果
2.1 α1A、α1B-AR引起的 [Ca2+ ]i变化 10 μmol.L-1 Adr激动α1A、α1B-AR后,能引起 HEK α1A和 HEKα1B 细胞[Ca2+ ]i的双相变化,包括起始的快速上升相(Ca2+释放相)和随后的持续平台相(Ca2+内流相)(见表1)。α1B-AR引起的Ca2+净释放量大于α1A-AR引起的Ca2+净释放量(P<0.01),两种受体亚型引起的Ca2+净内流量则差异无显著性(P>0.05)。
, http://www.100md.com
Tab 1 Change of [Ca2+ ]i (nmol.L-1) induced by 10 μmol.L-1 Adr in HEK α1A and HEK α1B cells
cells
n
resting
transient
plateau
[Ca2+] of influx
[Ca2+] of release
, http://www.100md.com
HEKα1A
10
96.7±27.4
245.6±66.3**
151.6±30.1
54.9±23.9
148.9±59.9**
HEKα1B
15
63.4±29.7
570.8±223.5
, 百拇医药
140.5±30.9
71.2±35.2
507.4±237.5
**P<0.01 vs HEKα1B. The n represented the number of determination
2.2 呋塞米和SK&F96365对α1A 、α1B-AR引起的Ca2+内流的影响 10 μmol.L-1 Adr可使HEKα1A 和HEKα1B细胞的静息[Ca2+]i从(102.8±28.1),(66.9±23.1) nmol.L-1升至(291.0±77.6),(560.1±286.2)nmol.L-1,然后逐渐到达平台相,此时[Ca2+]i为(152.3±39.3),(158.3±25.0) nmol.L-1(n=5; n为实验的例数,下同)。Cl-通道阻断剂呋塞米可呈浓度依赖性地降低平台相的[Ca2+]i (P<0.05, 见表2)。在同一剂量下,呋塞米对2种α1-AR亚型引起的Ca2+内流的抑制作用差异无显著性(P>0.05)。
, 百拇医药
Tab 2 Effects of furosemide and SK&F96365 on Adr-induced Ca2+ influx (nmol.L-1) in HEKα1A and HEKα1B cells
HEKα1A cells
HEKα1B cells
[Ca2+] of influx
Inhibition/%
[Ca2+]of influx
Inhibition/%
, 百拇医药
Pretreatment
49.7±20.4(5)
-
108.0±30.4(5)
-
Furosemide/μmol.L-1
2.5
45.4±19.0(4)
8.3±6.1*
95.8±31.0(4)
12.0±5.4*
, 百拇医药
5.0
36.0±10.7(4)
23.8±14.8*
88.1±28.6(4)
19.2±4.9*
10.0
29.6±14.6(5)
40.7±19.3*
73.7±22.9(5)
31.9±4.9*
pretreatment
, 百拇医药
64.0±26.4(5)
-
85.8±31.5(5)
-
SK&F96365/μmol.L-1
5.0
57.2±25.8(5)
10.6±6.4*
83.5±34.1(4)
8.1±5.7*
10.0
, http://www.100md.com
53.2±24.7(5)
16.6±9.6*
73.3±33.1(4)
19.9±9.8*
20.0
37.7±23.2(5)
43.8±14.6*
60.1±29.0(5)
32.1±8.1*
*P<0.05 vs pretreatment. The number of determination was shown in parentheses
, 百拇医药
在另一组实验中,10 μmol.L-1 Adr可使HEKα1A 和HEKα1B细胞的静息[Ca2+]i 从(91.8±7.2),(36.7±8.9) nmol.L-1升至(218.6±43.1),(651.9±206.6) nmol.L-1,然后逐渐到达平台相,此时[Ca2+]i为(161.0±12.8),(122.6±26.3) nmol.L-1(n=4)。ROC通道阻断剂SK&F96365可呈浓度依赖性地降低平台相的[Ca2+]i ,并在20 μmol.L-1时达到最大抑制作用 (P<0.05, 见表2)。在同一剂量下, SK&F96365对2种α1-AR亚型引起的Ca2+内流的抑制作用差异无显著性(P>0.05)。
, 百拇医药
2.3 呋塞米对SK&F96365抑制后的Ca2+内流的影响 10 μmol.L-1 Adr可使HEKα1A细胞的静息[Ca2+]i从(97.1±29.8) nmol.L-1升至(218.6±43.1) nmol.L-1,然后逐渐到达平台相,此时[Ca2+]i为(161.0±12.8) nmol.L-1(n=4)。20 μmol.L-1 SK&F96365可使平台相的[Ca2+]i 降低至(123.4±29. 9)nmol.L-1 ,抑制率达47.0%±12.1%。此时加入10 μmol.L-1呋塞米,不能进一步降低[Ca2+]i(P>0.05,n=4,见图1)。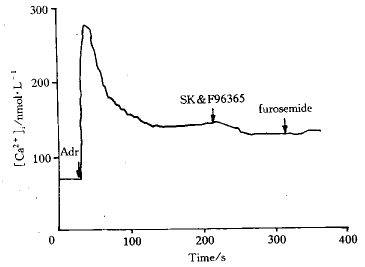
, 百拇医药
Fig 1 The effects of 10 μmol.L-1 furosemide and μmol.L-1 SK&F96365 on 10 μmol.L-1 Adr-induced Ca2+ influx i n HEKα1A cells
When SK&F96365 inhibited the Ca2+ influx by a maximal extent, furosemide d id not further decrease the [Ca2+] level
10 μmol.L-1 Adr同样可使HEKα1B细胞的静息[Ca2+ ]i从(36.7±8.9) nmol.L-1升至(651.9±206.6) nmol.L-1,然后稳定在(122.6±26.3) nmol.L-1水平(n=4)。在平台相加入20 μmol.L-1 SK&F96365可使 [Ca2+]i降低至(96.9±23.9) nmol.L-1,抑制率达32.1%±8.1%。此时加入10 μmol.L-1呋塞米,可使[Ca2+]i进一步下降至(87.6±23.8) nmol.L-1,抑制率达6.6%±3.2%(P<0.05,n=4,见图2)。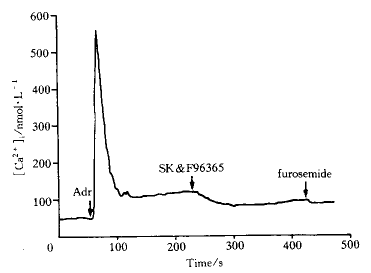
, 百拇医药
Fig 2 The effects of 10 μmol.L-1 furosemide and 20 μmol.L-1 SK&F96365 on 10 μmol.L-1 Adr-induced Ca2+ influ x in HEKα1B cells
When SK&F96365 inhibited the Ca2+ influx by a maximal extent, furosemide d id not further decrease the [Ca2+] level
2.4 SK&F96365对呋塞米抑制后的Ca2+内流的影响 10 μmol.L-1 Adr可使HEKα 1A细胞的静息[Ca2+ ]i从(91.8±7.2) nmol.L-1升至(280.3± 24.6) nmol.L-1 ,然后逐渐到达平台相,此时[Ca2+]i为(152.3 ±39.3) nmol.L-1(n=4)。10 μmol.L-1 呋塞米可使平台相的[Ca 2+ ]i降低至(132.1±32.0) nmol.L-1,抑制率达40.7%±19.4% 。此时加入20 μmol.L-1 SK&F96365,可使 [Ca2+]i进一步下降至 (127.4±31.9) nmol.L-1,抑制率达9.75%±3.01%(P<0.05,n=4) 。
, 百拇医药
10 μmol.L-1 Adr同样可使HEKα1B细胞的静息[Ca2+ ]i从(86.4±30.3)nmol.L-1升至(500.4±188.9) nmol.L-1,然后稳定在(14 0.8±35.0)nmol.L-1水平(n=5)。在平台相加入10 μmol.L-1呋塞 米可使 [Ca2+ ]i降低至(123.8±35.0) nmol.L-1 ,抑制率达35.5%±9.1%。此时加入20 μmol.L-1 SK&F96365,可使 [Ca2+ ]i进一步下降至(118.1±34.9) nmol.L-1,抑制率达8.4%±4.4%(P<0.05,n=5)。 HEKα1A 和HEKα1B 细胞上,SK&F96365对呋塞米抑制后的Ca2+内流的影响差异无显著性(P>0.05)。
, 百拇医药
2.5 硝苯地平对α1A、α1B激动引起的Ca2+内流的影响 10 μmol.L-1 Adr可使HEKα1A细胞的静息[Ca2+ ]i从(97.1±29.8)nmol.L-1升至(212.6±43.1)nmol.L-1,然后稳定在(161.0±12.8)nmol.L-1水平(n=5)。此时加入1 μmol.L-1 VDC通道阻断剂硝苯地平,[Ca2+ ]i并不进一步下降(P>0.05)。
同样,10 μmol.L-1Adr可使HEKα1B细胞的静息[Ca2+ ]i从(79.2±35.0)nmol.L-1升至(579.8±91.6)nmol.L-1,然后稳定在(157.5±23.7)nmol.L-1水平(n=5)。此时加入1 μmol.L-1硝苯地平,[Ca2+ ]i并不进一步下降(P>0.05)。
, 百拇医药
3 讨论
表1的结果表明,α1A和α1B-AR引起的Ca2+释放量是不同的,这与Tao[7] 关于α1B-AR产生的Ca2+释放要大于α1A-AR产生的Ca 2+释放的报道是一致的。可能是不同的α1-AR亚型涉及了胞内不同的Ca2+ 池释放所致。
虽然α1A和α1B-AR引起的Ca2+内流水平并无显著性差异,但是这两种内流的组成可能是不同的。使用Cl-通道阻断剂呋塞米,可以呈浓度依赖性地抑制这两 种α1-AR亚型引起的 Ca2+内流,说明其中有Cl-通道的参与;但是参与这两种 α1-AR亚型引起的Ca2+内流的Cl-通道种类可能是不同的。尽管ROC通道阻断剂S K&F96365对它们有相似的抑制作用,然而当SK&F96365达到最大抑制作用后,10 μmol.L-11呋塞米能进一步抑制α1B-AR引起的Ca2+内流,但不能进一步抑制α 1A-AR引起的Ca2+内流。这提示了至少有两种Cl-通道的开放参与α1A 和α1B-AR引起的Ca2+内流。一种是呋塞米和SK&F96365都敏感的,另一种是 呋塞米敏感而SK&F96365不敏感的。已经知道,Cl-通道具有多种类型[8],而受 体引起Cl-通道开放的程度则与Ca2+释放密切相关[6,9]。我们已经证明, 在HEKα1A和HEKα1B细胞上,α1B -AR引起的Ca2+释放涉及CPA ( Ca2+泵抑制剂)敏感及Adr敏感的两种Ca2+池,而α1A-AR引起的Ca 2+释放仅涉及Adr敏感的Ca2+池,与CPA敏感的Ca2+池无关[7] 。因此α1A和α1B-AR可能是通过引起不同的Ca2+池释放而引起了不同 的Cl-通道开放。
, 百拇医药
应该指出,同一剂量的呋塞米对α1A和α1B-AR引起的Ca2+内流的影响并无显著差异; 当使用10 μmol.L-1 呋塞米抑制这两种α1-AR亚型引起的Ca2+内流后,20 μmol.L-1 SK&F96365仍能产生进一步的抑制,彼此也没有显著性差异。可能的解释是,对比于α1B-AR和α1A-AR引起的Ca2+内流 中,呋塞米和SK&F96365都敏感的Cl-通道参与的程度要大一些。由于α1B-AR引起 的Ca2+内流中还涉及少量呋塞米敏感而SK&F96365不敏感的Cl-通道开放,因此总体 上这两种Ca2+内流中呋塞米敏感的Cl-通道开放参与的程度是相似的。
, 百拇医药
既往的研究认为,在血管平滑肌细胞上,α1-AR激动后,产生Ca2+释放,激活Ca2+依赖性的Cl-通道开放,引起Cl-外流,细胞膜去极化,进而引起VDC通道开放,产生持续性的Ca2+内流[10,11]。我们的研究表明,VDC通道阻断剂硝苯 地平对上述两种Ca2+内流没有影响,即HEK293细胞上α1A和α1B-AR引 起的Ca2+内流并无VDC通道的参与。这提示了在不同特性的细胞上,Cl-通道参与Ca 2+信号转导的机制是不同的;而在HEK293细胞上Cl-通道开放是如何调节ROC通道开 放的,尚需进一步研究。
我们的结果表明,在HEK293转染细胞上, α1A和α1B-AR引起的[Ca2+]i变化,不仅Ca2+释放相不同,而且Ca2+内流的组分也有差异,可能有不同数量和/(或)种类的Cl-通道参与。
, 百拇医药
*国家自然科学基金(No 39770858)和香港求是科技基金资助课题
作者简介:李劲梁,男,27岁,博士研究生;
关永源,男,教授,博士生导师,现为国务院学位委员会学科评议组成员;
韩启德,男,教授,博士生导师,中国科学院院士
李劲梁(中山医科大学药理学教研室,广州 510089)
关永源(中山医科大学药理学教研室,广州 510089)
韩启德(北京医科大学第三医院血管医学研究所,北京 100083)
贺华(中山医科大学药理学教研室,广州 510089)
, http://www.100md.com
丘钦英(中山医科大学药理学教研室,广州 510089)
参考文献
1,Marty A. The physiological role of calcium-dependent channels. Trends Neurosci, 1989; 12:420~4
2,Berridge MJ. Capacitative calcium entry. Biochem J, 1995;312:1~11
3,Gray LS, Russell JH. Cytolytic T lymphocytes effector function r equires plasma membrane chloride flux.J Immunol,1986;136:30 32~7
, 百拇医药
4,Rosoff PM, Hall C, Gramates LS, Terlecky SR.4,4-diisothiocyanatostlbene-2,2-disulfonic acid inhibits CD3-T cell antigen receptor-stimulated Ca2+influx in human T lymphocytes.J Biol Chem,1988;263(36):19535~40
5,Gericke M, Oike M, Droogmans G, Nilius B. Inhibition of capacitative Ca2+entry by Cl- channel blocker in human endothelial cells. Eur J Pharmacol, 1994;269:381~4
6,Amedee T, Large WA, Wang Q. Charateristics of chloride currents activated by noradrenaline in rabbit ear artery cells. J Physiol, 1990;428:501~18
, 百拇医药
7,Tao L, Guan YY, He H et al. Comparison of the Ca2+ movement by activation of alpha1- adrenoceptor subtypes in HEK-293 cells. Life Sci,1997;61(21):2127~36
8,Kuriyama H, Kitamura K, Nabata H. Pharmacological and physiolo gical significance of ion channels and factors that modulate them in vascular ti ssues. Pharmacol Rev, 1995;47:387~573
9,Lamb FS, Volk KA, Shibata EF. Calcium-activated chloride current in rabbit coronary artery myocytes.Circul Res,1994;75:742 ~50
, 百拇医药
10,Klockner U. Intracellular calcium ions activate a low-conductance chloride channel in smoothmuscle cells isolated from human mesenteric artery.Pfl ugers Arch, 1993;424:231~7
11,White CR, Elton TS, Shoemaker RL, Brock TA. Calcium-sensitive chloride channels in vascular smooth muscle cells. Proc Soc Exp Biol Med, 1995;208:255~62, http://www.100md.com
摘 要 目的 探讨Cl-通道开放在不同α1肾上腺素受体亚型引起的Ca2+内流中的作用。方法 采用Fura-2/AM荧光分光光度法,测定胞浆游离Ca2+浓度,观察Cl-通道阻断剂呋塞米,钙通道阻断剂SK&F96365和硝苯地平对不同α1肾上腺素受体亚型(α1A、α1B-AR)引起的Ca2+内流的影响,并加以比较。结果 2.5,5和10 μmol.L-1呋塞米呈浓度依赖性地抑制了α1A和α1B-AR亚型引起的Ca2+内流;对α1A-AR引起的Ca2+内流的抑制率分别为8.3%±6.1%,23.8%±14.8%和40.7%±19.3%,对α1B-AR引起的Ca2+内流的抑制率分别为12.0%±5.4%,19.2%±4.9%和31.9%±4.9%。α1A-AR引起的Ca2+内流被最大抑制浓度(20 μmol.L-1)的SK&F96365抑制后,不能被10 μmol.L-1呋塞米进一步抑制;而α1B-AR引起的Ca2+内流被20 μmol.L-1 SK&F96365抑制后,仍能被10 μmol.L-1呋塞米进一步抑制,抑制率为6.6%±3.2%。结论 参与α1A和α1B-AR引起的Ca2+内流的Cl-通道是非同一性的。
, 百拇医药
关键词:α1肾上腺素受体 Ca2+内流 Cl-通道 呋塞米 SK&F96365 硝苯地平
细胞内游离Ca2+水平的升高是许多生理、病理过程的起始信号,象血管的收缩,细胞的分泌,以及某些疾病如高血压、动脉粥样硬化等的发生都与之密切相关[1]。而受体介导的持续性Ca2+内流(re-ceptor-operated-Ca2+-influx;ROC)则是引起胞浆Ca2+水平升高的重要形式[2]。受体激动后,通过活化磷酯酰肌醇系统,水解出IP3,引起细胞内Ca2+池的Ca2+释放,进而触发因Ca2+释放引起的Ca2+内流(Ca2+-release-activated-Ca2+-influx; CRAC)。但是,Ca2+释放是通过什么机制引起Ca2+内流的,目前尚不清楚。Cl-离子是细胞外液中的主要阴离子。近来的研究表明,Cl-通道的开放对于CRAC的产生具有重要的意义[3~5]。在外周血管平滑肌的实验表明,Cl-通道参与了α1肾上腺素受体(α1-AR)激活引起的Ca2+内流[6]。神经递质去甲肾上腺素(noradrenaline;NA)激动α1-AR后,引起Ca2+释放,胞浆Ca2+水平升高,导致Cl-通道的开放,后者能进一步引起胞膜Ca2+通道的开放,产生持续性的Ca2+内流。选择性的α1-AR阻断剂哌唑嗪和一些Cl-通道阻断剂(如DIDS,呋塞米等)能不同程度地抑制Ca2+释放激活的Cl-通道开放[6]。本实验室的研究已经证实,参与ROC和电压依赖性的Ca2+内流(voltage-depedent- Ca2+-influx;VDC)的Cl-通道是非同一性的(未发表)。不同的α1-AR亚型引起的Ca2+内流中,参与的Cl-通道是否也存在这种情况,还有待进一步研究。
, 百拇医药
本研究以转染了α1A、α1B-AR cDNA的HEK293细胞为模型,采用Fura-2/AM荧光探针,测定胞浆游离Ca2+浓度([Ca2+]i),观察Cl- 通道阻断剂呋塞米及ROC通道阻断剂SK&F96365和VDC通道阻断剂硝苯地平对α1A、α1B-AR引起的Ca2+内流的影响,并加以比较。
1 材料与方法
1.1 实验材料 用牛α1A-AR和仓鼠α1B-AR cDNA转染的HEK293细胞(分别定义为HEKα1A和HEKα1B细胞)均由Dr. Minneman馈赠。Fura-2/AM,牛血清白蛋白(BSA),histidinol, hygromycin购于Borhringer Mannheim公司。肾上腺素(adrenaline;Adr),呋塞米(furosemide),硝苯地平(nifedipine), triton X-100, EGTA, HEPES购于Sigma公司。SK&F96365 购于Biomol 公司。DMEM/F12购于Gibico公司。小牛血清由本校微生物教研室提供。其余试剂均为国产分析纯。
, http://www.100md.com
1.2 实验仪器 RF-5 000荧光分光光度计(日本岛津公司),CO2培养箱(美国Precision公司),倒置显微镜(重庆光学仪器厂)。
1.3 实验方法
1.3.1 细胞培养 HEK293细胞用含10%(V/V)的小牛血清,100×103 U.L-1青霉素和125 mg.L-1链霉素的DMLE M/F12培养基,置于5% CO2,37℃的细胞培养箱内培养。HEKα1A和HEK1B细 胞分别用含有0.60 g.L-1 histidinol和0.05 g.L-1 hygromycin筛选抗生 素的培养基(含10%小牛血清的DMEM/F12)在上述条件下培养。细胞传代时,先用PBS(mmol.L -1: NaCl 130.0, KCl 2.5,Na2HPO4 10.0, KH2PO4 1.5,pH 7.4)洗1 次,加消化液(0.05% trypsin和0.02% EDTA溶于PBS,pH 7.4)消化30 s,吸弃消化液, 加入DMEM/F12终止消化,吹打后接种传代。
, 百拇医药
1.3.2 细胞[Ca2+]i测定 按本实验室已建立的方法进行[7],简述如下:当细胞生长至70%~ 90%融合时收获。用消化液消化细胞后,加入DMEM/F12(含0.05% BSA)终止消化。离心后 弃去上清,用含1 μmol.L-1 Fura-2/AM的DMEM/F12 (含0.05% BSA)重悬细胞,置 37℃、5% CO2条件下温育30 min。离心洗去细胞外的Fura-2/AM ,用BSS缓冲液(mmol.L -1: NaCl 130.0,KCl 5.0,MgCl2 1.0,CaCl2 1.5,HEPES 20.0, Glucose 10 .0, BSA 1.0 g, pH 7.4)重悬细胞,调整细胞密度至3×106个细胞/ml,在37℃恒温、磁力搅拌下进行荧光测定细胞[Ca2+]i。测定所用激发光波长为340 nm和380 nm,发射光波长为510 nm;测定结果由计算机自动记录,计算,并打印。所有实验试剂在有效剂量范围内都经证明无荧光干扰。
, 百拇医药
1.4 统计方法 实验结果以±s表示;组间及给药前后的[Ca2+]i差异用组间或配对t检验;P<0.05认为在统计学上差异有显著性。Adr引起的Ca2+净释放量和Ca2+净内流量及药物对Ca2+内流的抑制率按下列公式计算:
Ca2+净释放量([Ca2+] of release)=快速上升相[Ca2+]i-静息[Ca2+]i
Ca2+净内流量([Ca2+] of influx)=平台相[Ca2+]i-静息[Ca2+]i
, http://www.100md.com 抑制率(%)=(加药前平台相[Ca2+]i-加药后平台相[Ca2+]i)/(加药前平台相[Ca2+]i-静息[Ca2+]i)×100%
2 结果
2.1 α1A、α1B-AR引起的 [Ca2+ ]i变化 10 μmol.L-1 Adr激动α1A、α1B-AR后,能引起 HEK α1A和 HEKα1B 细胞[Ca2+ ]i的双相变化,包括起始的快速上升相(Ca2+释放相)和随后的持续平台相(Ca2+内流相)(见表1)。α1B-AR引起的Ca2+净释放量大于α1A-AR引起的Ca2+净释放量(P<0.01),两种受体亚型引起的Ca2+净内流量则差异无显著性(P>0.05)。
, http://www.100md.com
Tab 1 Change of [Ca2+ ]i (nmol.L-1) induced by 10 μmol.L-1 Adr in HEK α1A and HEK α1B cells
cells
n
resting
transient
plateau
[Ca2+] of influx
[Ca2+] of release
, http://www.100md.com
HEKα1A
10
96.7±27.4
245.6±66.3**
151.6±30.1
54.9±23.9
148.9±59.9**
HEKα1B
15
63.4±29.7
570.8±223.5
, 百拇医药
140.5±30.9
71.2±35.2
507.4±237.5
**P<0.01 vs HEKα1B. The n represented the number of determination
2.2 呋塞米和SK&F96365对α1A 、α1B-AR引起的Ca2+内流的影响 10 μmol.L-1 Adr可使HEKα1A 和HEKα1B细胞的静息[Ca2+]i从(102.8±28.1),(66.9±23.1) nmol.L-1升至(291.0±77.6),(560.1±286.2)nmol.L-1,然后逐渐到达平台相,此时[Ca2+]i为(152.3±39.3),(158.3±25.0) nmol.L-1(n=5; n为实验的例数,下同)。Cl-通道阻断剂呋塞米可呈浓度依赖性地降低平台相的[Ca2+]i (P<0.05, 见表2)。在同一剂量下,呋塞米对2种α1-AR亚型引起的Ca2+内流的抑制作用差异无显著性(P>0.05)。
, 百拇医药
Tab 2 Effects of furosemide and SK&F96365 on Adr-induced Ca2+ influx (nmol.L-1) in HEKα1A and HEKα1B cells
HEKα1A cells
HEKα1B cells
[Ca2+] of influx
Inhibition/%
[Ca2+]of influx
Inhibition/%
, 百拇医药
Pretreatment
49.7±20.4(5)
-
108.0±30.4(5)
-
Furosemide/μmol.L-1
2.5
45.4±19.0(4)
8.3±6.1*
95.8±31.0(4)
12.0±5.4*
, 百拇医药
5.0
36.0±10.7(4)
23.8±14.8*
88.1±28.6(4)
19.2±4.9*
10.0
29.6±14.6(5)
40.7±19.3*
73.7±22.9(5)
31.9±4.9*
pretreatment
, 百拇医药
64.0±26.4(5)
-
85.8±31.5(5)
-
SK&F96365/μmol.L-1
5.0
57.2±25.8(5)
10.6±6.4*
83.5±34.1(4)
8.1±5.7*
10.0
, http://www.100md.com
53.2±24.7(5)
16.6±9.6*
73.3±33.1(4)
19.9±9.8*
20.0
37.7±23.2(5)
43.8±14.6*
60.1±29.0(5)
32.1±8.1*
*P<0.05 vs pretreatment. The number of determination was shown in parentheses
, 百拇医药
在另一组实验中,10 μmol.L-1 Adr可使HEKα1A 和HEKα1B细胞的静息[Ca2+]i 从(91.8±7.2),(36.7±8.9) nmol.L-1升至(218.6±43.1),(651.9±206.6) nmol.L-1,然后逐渐到达平台相,此时[Ca2+]i为(161.0±12.8),(122.6±26.3) nmol.L-1(n=4)。ROC通道阻断剂SK&F96365可呈浓度依赖性地降低平台相的[Ca2+]i ,并在20 μmol.L-1时达到最大抑制作用 (P<0.05, 见表2)。在同一剂量下, SK&F96365对2种α1-AR亚型引起的Ca2+内流的抑制作用差异无显著性(P>0.05)。
, 百拇医药
2.3 呋塞米对SK&F96365抑制后的Ca2+内流的影响 10 μmol.L-1 Adr可使HEKα1A细胞的静息[Ca2+]i从(97.1±29.8) nmol.L-1升至(218.6±43.1) nmol.L-1,然后逐渐到达平台相,此时[Ca2+]i为(161.0±12.8) nmol.L-1(n=4)。20 μmol.L-1 SK&F96365可使平台相的[Ca2+]i 降低至(123.4±29. 9)nmol.L-1 ,抑制率达47.0%±12.1%。此时加入10 μmol.L-1呋塞米,不能进一步降低[Ca2+]i(P>0.05,n=4,见图1)。
, 百拇医药
Fig 1 The effects of 10 μmol.L-1 furosemide and μmol.L-1 SK&F96365 on 10 μmol.L-1 Adr-induced Ca2+ influx i n HEKα1A cells
When SK&F96365 inhibited the Ca2+ influx by a maximal extent, furosemide d id not further decrease the [Ca2+] level
10 μmol.L-1 Adr同样可使HEKα1B细胞的静息[Ca2+ ]i从(36.7±8.9) nmol.L-1升至(651.9±206.6) nmol.L-1,然后稳定在(122.6±26.3) nmol.L-1水平(n=4)。在平台相加入20 μmol.L-1 SK&F96365可使 [Ca2+]i降低至(96.9±23.9) nmol.L-1,抑制率达32.1%±8.1%。此时加入10 μmol.L-1呋塞米,可使[Ca2+]i进一步下降至(87.6±23.8) nmol.L-1,抑制率达6.6%±3.2%(P<0.05,n=4,见图2)。
, 百拇医药
Fig 2 The effects of 10 μmol.L-1 furosemide and 20 μmol.L-1 SK&F96365 on 10 μmol.L-1 Adr-induced Ca2+ influ x in HEKα1B cells
When SK&F96365 inhibited the Ca2+ influx by a maximal extent, furosemide d id not further decrease the [Ca2+] level
2.4 SK&F96365对呋塞米抑制后的Ca2+内流的影响 10 μmol.L-1 Adr可使HEKα 1A细胞的静息[Ca2+ ]i从(91.8±7.2) nmol.L-1升至(280.3± 24.6) nmol.L-1 ,然后逐渐到达平台相,此时[Ca2+]i为(152.3 ±39.3) nmol.L-1(n=4)。10 μmol.L-1 呋塞米可使平台相的[Ca 2+ ]i降低至(132.1±32.0) nmol.L-1,抑制率达40.7%±19.4% 。此时加入20 μmol.L-1 SK&F96365,可使 [Ca2+]i进一步下降至 (127.4±31.9) nmol.L-1,抑制率达9.75%±3.01%(P<0.05,n=4) 。
, 百拇医药
10 μmol.L-1 Adr同样可使HEKα1B细胞的静息[Ca2+ ]i从(86.4±30.3)nmol.L-1升至(500.4±188.9) nmol.L-1,然后稳定在(14 0.8±35.0)nmol.L-1水平(n=5)。在平台相加入10 μmol.L-1呋塞 米可使 [Ca2+ ]i降低至(123.8±35.0) nmol.L-1 ,抑制率达35.5%±9.1%。此时加入20 μmol.L-1 SK&F96365,可使 [Ca2+ ]i进一步下降至(118.1±34.9) nmol.L-1,抑制率达8.4%±4.4%(P<0.05,n=5)。 HEKα1A 和HEKα1B 细胞上,SK&F96365对呋塞米抑制后的Ca2+内流的影响差异无显著性(P>0.05)。
, 百拇医药
2.5 硝苯地平对α1A、α1B激动引起的Ca2+内流的影响 10 μmol.L-1 Adr可使HEKα1A细胞的静息[Ca2+ ]i从(97.1±29.8)nmol.L-1升至(212.6±43.1)nmol.L-1,然后稳定在(161.0±12.8)nmol.L-1水平(n=5)。此时加入1 μmol.L-1 VDC通道阻断剂硝苯地平,[Ca2+ ]i并不进一步下降(P>0.05)。
同样,10 μmol.L-1Adr可使HEKα1B细胞的静息[Ca2+ ]i从(79.2±35.0)nmol.L-1升至(579.8±91.6)nmol.L-1,然后稳定在(157.5±23.7)nmol.L-1水平(n=5)。此时加入1 μmol.L-1硝苯地平,[Ca2+ ]i并不进一步下降(P>0.05)。
, 百拇医药
3 讨论
表1的结果表明,α1A和α1B-AR引起的Ca2+释放量是不同的,这与Tao[7] 关于α1B-AR产生的Ca2+释放要大于α1A-AR产生的Ca 2+释放的报道是一致的。可能是不同的α1-AR亚型涉及了胞内不同的Ca2+ 池释放所致。
虽然α1A和α1B-AR引起的Ca2+内流水平并无显著性差异,但是这两种内流的组成可能是不同的。使用Cl-通道阻断剂呋塞米,可以呈浓度依赖性地抑制这两 种α1-AR亚型引起的 Ca2+内流,说明其中有Cl-通道的参与;但是参与这两种 α1-AR亚型引起的Ca2+内流的Cl-通道种类可能是不同的。尽管ROC通道阻断剂S K&F96365对它们有相似的抑制作用,然而当SK&F96365达到最大抑制作用后,10 μmol.L-11呋塞米能进一步抑制α1B-AR引起的Ca2+内流,但不能进一步抑制α 1A-AR引起的Ca2+内流。这提示了至少有两种Cl-通道的开放参与α1A 和α1B-AR引起的Ca2+内流。一种是呋塞米和SK&F96365都敏感的,另一种是 呋塞米敏感而SK&F96365不敏感的。已经知道,Cl-通道具有多种类型[8],而受 体引起Cl-通道开放的程度则与Ca2+释放密切相关[6,9]。我们已经证明, 在HEKα1A和HEKα1B细胞上,α1B -AR引起的Ca2+释放涉及CPA ( Ca2+泵抑制剂)敏感及Adr敏感的两种Ca2+池,而α1A-AR引起的Ca 2+释放仅涉及Adr敏感的Ca2+池,与CPA敏感的Ca2+池无关[7] 。因此α1A和α1B-AR可能是通过引起不同的Ca2+池释放而引起了不同 的Cl-通道开放。
, 百拇医药
应该指出,同一剂量的呋塞米对α1A和α1B-AR引起的Ca2+内流的影响并无显著差异; 当使用10 μmol.L-1 呋塞米抑制这两种α1-AR亚型引起的Ca2+内流后,20 μmol.L-1 SK&F96365仍能产生进一步的抑制,彼此也没有显著性差异。可能的解释是,对比于α1B-AR和α1A-AR引起的Ca2+内流 中,呋塞米和SK&F96365都敏感的Cl-通道参与的程度要大一些。由于α1B-AR引起 的Ca2+内流中还涉及少量呋塞米敏感而SK&F96365不敏感的Cl-通道开放,因此总体 上这两种Ca2+内流中呋塞米敏感的Cl-通道开放参与的程度是相似的。
, 百拇医药
既往的研究认为,在血管平滑肌细胞上,α1-AR激动后,产生Ca2+释放,激活Ca2+依赖性的Cl-通道开放,引起Cl-外流,细胞膜去极化,进而引起VDC通道开放,产生持续性的Ca2+内流[10,11]。我们的研究表明,VDC通道阻断剂硝苯 地平对上述两种Ca2+内流没有影响,即HEK293细胞上α1A和α1B-AR引 起的Ca2+内流并无VDC通道的参与。这提示了在不同特性的细胞上,Cl-通道参与Ca 2+信号转导的机制是不同的;而在HEK293细胞上Cl-通道开放是如何调节ROC通道开 放的,尚需进一步研究。
我们的结果表明,在HEK293转染细胞上, α1A和α1B-AR引起的[Ca2+]i变化,不仅Ca2+释放相不同,而且Ca2+内流的组分也有差异,可能有不同数量和/(或)种类的Cl-通道参与。
, 百拇医药
*国家自然科学基金(No 39770858)和香港求是科技基金资助课题
作者简介:李劲梁,男,27岁,博士研究生;
关永源,男,教授,博士生导师,现为国务院学位委员会学科评议组成员;
韩启德,男,教授,博士生导师,中国科学院院士
李劲梁(中山医科大学药理学教研室,广州 510089)
关永源(中山医科大学药理学教研室,广州 510089)
韩启德(北京医科大学第三医院血管医学研究所,北京 100083)
贺华(中山医科大学药理学教研室,广州 510089)
, http://www.100md.com
丘钦英(中山医科大学药理学教研室,广州 510089)
参考文献
1,Marty A. The physiological role of calcium-dependent channels. Trends Neurosci, 1989; 12:420~4
2,Berridge MJ. Capacitative calcium entry. Biochem J, 1995;312:1~11
3,Gray LS, Russell JH. Cytolytic T lymphocytes effector function r equires plasma membrane chloride flux.J Immunol,1986;136:30 32~7
, 百拇医药
4,Rosoff PM, Hall C, Gramates LS, Terlecky SR.4,4-diisothiocyanatostlbene-2,2-disulfonic acid inhibits CD3-T cell antigen receptor-stimulated Ca2+influx in human T lymphocytes.J Biol Chem,1988;263(36):19535~40
5,Gericke M, Oike M, Droogmans G, Nilius B. Inhibition of capacitative Ca2+entry by Cl- channel blocker in human endothelial cells. Eur J Pharmacol, 1994;269:381~4
6,Amedee T, Large WA, Wang Q. Charateristics of chloride currents activated by noradrenaline in rabbit ear artery cells. J Physiol, 1990;428:501~18
, 百拇医药
7,Tao L, Guan YY, He H et al. Comparison of the Ca2+ movement by activation of alpha1- adrenoceptor subtypes in HEK-293 cells. Life Sci,1997;61(21):2127~36
8,Kuriyama H, Kitamura K, Nabata H. Pharmacological and physiolo gical significance of ion channels and factors that modulate them in vascular ti ssues. Pharmacol Rev, 1995;47:387~573
9,Lamb FS, Volk KA, Shibata EF. Calcium-activated chloride current in rabbit coronary artery myocytes.Circul Res,1994;75:742 ~50
, 百拇医药
10,Klockner U. Intracellular calcium ions activate a low-conductance chloride channel in smoothmuscle cells isolated from human mesenteric artery.Pfl ugers Arch, 1993;424:231~7
11,White CR, Elton TS, Shoemaker RL, Brock TA. Calcium-sensitive chloride channels in vascular smooth muscle cells. Proc Soc Exp Biol Med, 1995;208:255~62, http://www.100md.com