铁粒幼细胞贫血家系一个新的ALAS2基因突变
作者:朱平 卜定方
单位:100034 北京医科大学第一医院
关键词:贫血;基因,ALAS2;突变;聚合酶链反应
中华血液学杂志000909
【摘要】 目的 确定一个铁粒幼细胞贫血家系是由于ALAS2基因突变所致。方法 用聚合酶链反应扩增X染色体上与ALAS2距离小于1.7Mb的微卫星DXS 991和DXS 1199,用变性凝胶电泳进行3代家族成员9人(包括第2代2例患者)的单体型分析;克隆患者以及正常人ALAS2基因的cDNA全部编码区,并测序比较。结果 该家系2例先证者都从母亲获得同一ALAS2等位基因。健康兄、妹2人则获得另一条ALAS2等位基因。克隆患者和正常人ALAS2 cDNA编码区后测序发现,患者第5外显子A523G突变,导致苏氨酸变为丙氨酸;第3外显子T372C突变,导致亮氨酸由脯氨酸替代,后者在ALAS2可剪拼区内,意义尚不明确。结论 在X连锁遗传性铁粒幼细胞贫血家系新发现了一种导致发病的ALAS2基因第5外显子突变。
, 百拇医药
A novel mutation of the ALAS2 gene in a family with X-linked sideroblastic anemia
ZHU Ping BU Dingfang
(The First Teaching Hospital of Beijing Medical University, Beijing 100034,China)
【Abstract】 Objective To confirm the mutation of ALAS2 gene is the cause of sideroblastic anemia in a family. Methods Polymerase chain reaction (PCR) was used to amplify the microsatellite DXS 991, DXS 1199 in the chromosome Xp11.22 linked gene ALAS2 and haplotype analysis was performed in a kindred with 2 patients and 7 normal members. All cDNA encoded regions in the ALAS2 gene of the patients and their normal siblings were cloned, sequenced and compared. Result Both brother patients had the same allele of ALAS2 and their normal siblings did not. The mutation in the patients’ ALAS2 gene was exon 5 A523G, causing threonine to alanine; and exon 3 T372C, leucine to proline. The latter located in the splicing region, its significance is not clear. Conclusion The pathogenesis of this kindred of X-linked sideroblastic anemia (XLSA) involved a novel mutation in ALAS2 exon 5.
, 百拇医药
【Key word】 Anemia; Gene, ALAS2; Mutation; Polymerase chain reaction
铁粒幼细胞贫血(SA)包括一组铁利用障碍所致的贫血性疾病,患者由于骨髓红细胞系统生成障碍,出现严重程度不等的贫血,血清铁升高,骨髓出现大量铁颗粒,形成环形铁粒幼细胞。遗传性SA主要为X连锁性铁幼粒细胞贫血(XLSA)[1]。XLSA发病可能是由于红细胞系统特异性δ氨基γ酮戊酸合成酶2(ALAS2)基因突变所致[2]。我们最近发现一个3代铁粒幼细胞贫血家系,4兄妹中有2个兄弟发病。用与ALAS2距离小于1.7Mb的微卫星做连锁分析以了解该家系的遗传方式;克隆患者及正常人ALAS2基因的cDNA全编码区并测序。发现患者ALAS2基因存在一种新的突变,第5外显子A523G突变,导致苏氨酸变为丙氨酸。
对象和方法
, 百拇医药 1 病例
1.1 家系:先证者兄弟2人及其家系3代共9个成员,其中第1代1人,第2代6人,即先证者兄弟2人及其配偶1人,先证者之弟、妹2人及其配偶1人;第3代2人,其中之一为患者之子。均进行血常规、血清铁,铁蛋白、总铁结合力测定,血涂片检查,第1代母亲无贫血但红细胞呈双形性。第2代中患者兄弟2人均以糖尿病为首发症状,血清铁高,均在20岁左右出现低色素、小细胞贫血,骨髓可见大量环形铁粒幼细胞,血清铁明显增高。第2代另外4名成员和第3代2名男性后代正常。
1.2 先证者1:男,47岁。因“发现贫血27年,多饮、多尿、体重下降3个月”入院。27年前偶然发现贫血,按“缺铁性贫血”治疗数年无效。20年前始面色青灰。3个月前出现多饮、多尿、体重下降。空腹血糖3.9g/L。查体:全身皮肤呈青灰色,肝肋下2cm,质硬,边钝,表面光滑,压痛(+),脾肋缘下4~5cm,质硬,边钝,表面光滑,压痛(+),双下肢轻度可凹性水肿。血常规:Hb 59g/L,RBC 2.55×1012/L, WBC 2.7×109/L,BPC 236×109/L,网织0.016;骨髓象:骨髓增生明显活跃,红系52.5%,中幼红细胞增高,成熟红细胞明显大小不等,畸型红细胞多见,部分红细胞中心浅染区扩大,粒、红细胞数量比为0.7∶1,全片见38个巨核细胞;细胞外铁(++),细胞内铁阳性率0.63,其中Ⅰ型0.15,Ⅱ型0.02, Ⅲ型0.05,Ⅳ型0.23,环型0.18。铁蛋白>500μg/L,血清铁 48μmol/L,总铁结合力47μmol/L。馒头餐:血糖0,1,2,3h分别为:20.38, 24.71,28.50, 26.02mmol/L;胰岛素0,1,2,3h分别为:4.16,4.56,4.74,5.00mIU/L。T3 0.47mg/L (正常0.69~2.15mg/L),T4 39.8 mg/L (正常42.3~112.5mg/L),促甲状腺素(TSH)<0.03 mg/L (正常0.75~3.53mg/L)。尿17羟12.6mg/24h;尿17酮16.2mg/24h。促肾上腺皮质素(ACTH)114.44ng/L(正常6.0~56.7ng/L)。B超:肝大,肝实质回声致密,门脉主干宽1.4cm,脾大,肋缘下平脐,下腹盆腔可见少量积液。超声心动图:各房室大小正常,二尖瓣前后叶稍增厚,轻度关闭不全,瓣膜活动正常,室壁不厚,运动正常。Rous试验四次(-);蔗糖水试验(-);Ham试验(-);CD59阳性率100%;Coombs试验(-)。血红蛋白电泳两次(-)。尿卟胆原(-),G6PD酶活性正常。入院后予诺和灵降血糖,但多次发生低血糖发作,曾心悸,心电图示快速房颤,10min后自行缓解。诊断为铁粒幼细胞贫血,继发性血色病,继发性糖尿病。2个月后因顽固性心律失常心衰死亡。
, 百拇医药
1.3 先证者2,男,38岁。因“发现贫血20 余年,发现血糖高4个月”入院。20余年前查体发现 “缺铁性贫血”,因口渴,多饮,尿量增多,血糖高入院。查体:面色苍白,稍铁青,肝、脾触诊不满意,双踝部可凹性水肿。实验室检查:馒头餐试验血糖呈典型糖尿病曲线,铁蛋白>500μg/L,血清铁>90μmol/L,总铁结合力32.1μmol/L,网织红细胞不高,Coombs试验(-),Rous试验(-)。骨髓象:骨髓增生活跃,粒系0.31,粒、红细胞数量比为0.6∶1;红系比例0.50,中幼红细胞0.32,晚幼红细胞0.17,有核红细胞胞体小,胞浆少,成熟红细胞大小不等明显,中心浅染区扩大,畸型红细胞及大红细胞多见。全片见1个巨核细胞。细胞外铁(-),细胞内铁阳性率0.90,其中Ⅰ型0.06,Ⅱ型0.18,Ⅲ型0.22,Ⅳ型0.12,环型0.32。骨髓活检:符合铁粒幼细胞贫血。MRI提示肝、脾、胰脏信号异常,脾大,考虑铁沉积。血红蛋白电泳(两次):正常。T3 0.59 mg/L,T4 35.6 mg/L, TSH 2.81 mg/L。尿3-甲氧-4羟苦杰仁酸(VMA) <5mg/24h;尿糖定量52.5g/24h;尿17羟14.0mg/24h, 尿17酮10.5 mg/24 h。超声心动图:左房、室扩大并二尖瓣关闭不全; 主动脉瓣轻度关闭不全,左室收缩功能降低。诊断为铁粒幼细胞贫血,继发性血色病、糖尿病。
, http://www.100md.com
2 标本 取外周血细胞用高盐沉淀法提取DNA做基因分析;10名正常人外周血DNA和骨髓作为微卫星多态性正常对照;从患者骨髓细胞和3个月龄人工流产正常胎儿肝脏提取总mRNA,poly(dT)引物制备cDNA,以克隆ALAS2的cDNA。
3 PCR引物设计与合成 从国际互联网www.genethon.fr基因信息库中查得3对微卫星引物,送赛百盛公司合成。其中包括染色体Xp11.21上与ALAS 2距离小于1.7Mb的微卫星DXS 991和DXS 1199以及位于Xp22.13的DXS 1226。ALAS2 cDNA克隆的引物参照Genebank x56352的序列设计,有义链: 5′-ACTAGGATCCGCAGCCATGCTGCTACAGTGC-3′,反义链:5′-CTAGAATTCAGGCATAGGTGGTGACATAC-3′。
4 PCR扩增微卫星 25μl反应缓冲液中含有:模板DNA 0.25ng,Mg离子2mmol/L,4×dNTP 各50μmol/L,引物各10pmol。 Taq聚合酶0.5U。PCR反应程序:94℃ 5min变性后,进行以下30个循环:93℃ 30s,56℃ 30s,72℃ 1min。之后72℃延伸10min。实验重复2次以上。取PCR扩增反应液4~8μl加入1~2倍含体积分数为95%甲酰胺的溴酚蓝上样缓冲液,在含7mol/L尿素的60g/L的聚丙烯酰胺凝胶上,300 V进行电泳,银染显色。
, 百拇医药
5 从先证者2的骨髓和胎肝细胞中克隆ALAS2 cDNA的全部编码区 用淋巴细胞分离液分离骨髓有核细胞。骨髓有核细胞和胎肝细胞总RNA提取采用Gibco/BRL公司的TRIZOL试剂盒,按照说明提取RNA。 逆转录用该公司的Superscript cDNA第一链合成试剂盒,以poly(dT)为引物,逆转录反应的总体系为20μl。PCR反应体系25μl,含逆转录反应产物cDNA 1μl(作为PCR反应模板),有义链和反义链引物各25pmol,94℃预变性5min; 循环温度为94℃变性1min,54℃退火1min,72℃延伸90s,共30个循环; 循环结束后继续于72℃延伸7min。反应产物用聚丙烯酰胺凝胶电泳,银染法鉴定。
6 先证者2和胎儿ALAS2基因片段进行克隆和限制性酶谱分析 将25μl PCR产物经酚提取,乙醇沉淀后溶于5μl TE中,用BamHⅠ和EcoRⅠ酶消化1h,再用低熔点琼脂糖凝胶纯化。定向克隆于质粒pcDNA3 BamHⅠ和EcoRⅠ位点中。选择有克隆产物的质粒,BamHⅠ+EcoRⅠ和BstⅠ消化后,根据片段长度做鉴定。
, 百拇医药
7 测序分析 用ABI测序仪分3段正反向测序,分析全部1800bp左右的序列。中段测序引物5′-CCCAACATTCTGAG-3′。
结果
1 正常人DNA微卫星DXS 991和DXS 1199以及DXS 1226片段多态性 从10名正常人采集的外周血提取DNA,然后进行了3个微卫星位点的分析,结果不同个体的微卫星DXS 991和DXS 1199以及DXS 1226片段长短不同,呈多态性,可用作单体型分析。
2 先证者家系遗传连锁分析结果 2例先证者DXS 991和DXS 1199的多态性相联锁,也与母亲DXS 991和DXS 1199两等位基因之一相一致。提示患病兄弟从母系获得同样的致病性ALAS2基因。
3 用RT-PCR克隆获得先证者和正常胎肝造血细胞ALAS2 cDNA全部编码区 患者cDNA克隆的pcDNA经限制性内切酶BamHⅠ和EcoRⅠ消化电泳后证实为1700bp序列;胎肝则获得1700bp和1600bp两条片段(图1)。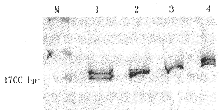
, 百拇医药
1,4:从胎肝获得1700和1600bp 2条ALAS2基因片段;
2,3:先证者获得1700bp 1条片段;M为分子量标志
图1 RT-PCR克隆ALAS2基因片段
4 克隆片段的限制性酶谱分析 先证者和胎儿的ALAS2/pcDNA3质粒均可被PstⅠ消化获得4500或4600,1385,595及540 bp 4条新的较小片段。与报道的ALAS2 cDNA相符(图2)。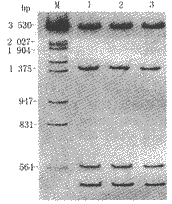
M:分子量标准λ DNA/EcoR Ⅰ+Hind Ⅲ; 1:病例2;
2:正常胎肝的1700bp片段; 3:正常胎肝的1600bp的片段
, http://www.100md.com
图2 ALAS2/pcDNA3克隆的PstⅠ酶切片段
5 经测序,从胎肝细胞获得的两ALAS2克隆,大片段与正常的ALAS2 cDNA序列(x60364)相同,小片段缺失第4外显子111 bp序列。
6 先证者与胎儿ALAS2序列对比分析 比较先证者和胎肝ALAS2发现,先证者第5外显子A523G突变(图3),导致苏氨酸变为丙氨酸;即ALAS2蛋白的T161A突变;第3外显子T372C突变,导致亮氨酸由脯氨酸替代,即ALAS2蛋白的L107P的突变。后者在ALAS2可剪拼区的第4外显子内。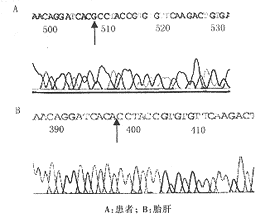
图3 测序发现先证者ALAS2基因第5外显子A523G突变
讨论
, http://www.100md.com SA是一组铁利用障碍所致的贫血性疾病。遗传性SA主要为X连锁SA(XLAS)。最近发现XLSA发病是由于红细胞线粒体中的δ氨基γ酮戊酸合成酶2(ALAS2)基因突变所致[2]。这些突变发生在第5~11外显子中,至1999年已有15种[3-5]。我们最近在北京地区发现一个XLSA家系,4兄妹中有2个兄弟发病,先用与ALAS2距离小于1.7Mb的微卫星DXS 991和DXS 1199以及距离较远、位于Xp22.13的DXS1226做连锁分析,两兄弟都从母系获得同样的DXS 991和DXS 1199基因。而患者距离较远的DXS 1226不连锁。表明该家系两患者兄弟都从母系获得同一ALAS2等位基因。健康兄、妹2人则获得另一条ALAS2等位基因。随后克隆患者和正常人ALAS2 cDNA编码区,经限制性酶谱和测序发现,患者第5外显子A523G突变,导致苏氨酸变为丙氨酸;这是国内外尚未报告过的一种新ALAS2基因突变, 也是首次在国内发现导致铁粒幼细胞贫血的基因突变。我们同时发现患者第4外显子T372C突变,可导致亮氨酸由脯氨酸替代,但这个突变位点在ALAS2 cDNA的可剪拼区内。Canboy等分析ALAS2基因发现其存在两种不同的异构体,一种异构体比另一种少37个氨基酸、111个碱基,缺失部分为ALAS2基因的第4外显子。他们认为这种缺失片段没有明显的功能差异。因此该突变可能不是该家族患病的致病基因位点,其意义有待进一步分析。从临床表现结合文献报道分析,这种X连锁的疾病有如下特征:往往在20岁左右发现贫血和症状;由于不适当地输血和补铁造成铁负荷过重,中年易出现继发性血色病,可并发糖尿病、肝硬化或者心肌病等;血清铁、铁蛋白明显增高;骨髓出现大量环形铁幼粒细胞。一些女性携带者可有脾肿大和少数红细胞异常,但是没有贫血。
, 百拇医药
基金项目:国家自然科学基金资助项目(3957321)和卫生部科研基金资助项目(98-2-2220)
参 考 文 献
1,Bottomley SS, Wise PD, Wassson EG, et al. X-linked sideroblastic anemia: update of molecular detects and mechanism of the associated iron overload. Acta Haematologica, 1997, 98:103.
2,Conboy JH, Cox TC, Bottemley S, et al. Human erythriod 5-aminolevulinate synthese gene structure and specific differences in alternative RNA splicing. J Bio Chem,1992, 267:18753-18758.
, 百拇医药
3,Cotter PD, May A, Li L, et al. Four new mutations in the erythroid-specific 5-aminolevulinate synthase (ALAS2) gene causing X-linked sideroblastic anemia: increased pyridoxine responsiveness after removal of iron overload by phlebotomy and coinheritance of hereditary hemochromatosis. Blood, 1999, 93: 1757-1769.
4,Cox TC, Bottomley SS, Wiley JS. et al. X-linked pyridoxine-responsive sideroblastic anemia due to a thr388-to-ser substitution in erythroid 5-aminolevulinate synthase. New Eng J Med, 1994. 330: 675-679.
5,Prades E, Chambon C, Dailey TA,et al. A new mutation of the ALAS2 gene in a large family with X-linked sideroblastic anemia. Hum Genet, 1995, 95: 424-428.
(收稿日期:1999-09-13), 百拇医药
单位:100034 北京医科大学第一医院
关键词:贫血;基因,ALAS2;突变;聚合酶链反应
中华血液学杂志000909
【摘要】 目的 确定一个铁粒幼细胞贫血家系是由于ALAS2基因突变所致。方法 用聚合酶链反应扩增X染色体上与ALAS2距离小于1.7Mb的微卫星DXS 991和DXS 1199,用变性凝胶电泳进行3代家族成员9人(包括第2代2例患者)的单体型分析;克隆患者以及正常人ALAS2基因的cDNA全部编码区,并测序比较。结果 该家系2例先证者都从母亲获得同一ALAS2等位基因。健康兄、妹2人则获得另一条ALAS2等位基因。克隆患者和正常人ALAS2 cDNA编码区后测序发现,患者第5外显子A523G突变,导致苏氨酸变为丙氨酸;第3外显子T372C突变,导致亮氨酸由脯氨酸替代,后者在ALAS2可剪拼区内,意义尚不明确。结论 在X连锁遗传性铁粒幼细胞贫血家系新发现了一种导致发病的ALAS2基因第5外显子突变。
, 百拇医药
A novel mutation of the ALAS2 gene in a family with X-linked sideroblastic anemia
ZHU Ping BU Dingfang
(The First Teaching Hospital of Beijing Medical University, Beijing 100034,China)
【Abstract】 Objective To confirm the mutation of ALAS2 gene is the cause of sideroblastic anemia in a family. Methods Polymerase chain reaction (PCR) was used to amplify the microsatellite DXS 991, DXS 1199 in the chromosome Xp11.22 linked gene ALAS2 and haplotype analysis was performed in a kindred with 2 patients and 7 normal members. All cDNA encoded regions in the ALAS2 gene of the patients and their normal siblings were cloned, sequenced and compared. Result Both brother patients had the same allele of ALAS2 and their normal siblings did not. The mutation in the patients’ ALAS2 gene was exon 5 A523G, causing threonine to alanine; and exon 3 T372C, leucine to proline. The latter located in the splicing region, its significance is not clear. Conclusion The pathogenesis of this kindred of X-linked sideroblastic anemia (XLSA) involved a novel mutation in ALAS2 exon 5.
, 百拇医药
【Key word】 Anemia; Gene, ALAS2; Mutation; Polymerase chain reaction
铁粒幼细胞贫血(SA)包括一组铁利用障碍所致的贫血性疾病,患者由于骨髓红细胞系统生成障碍,出现严重程度不等的贫血,血清铁升高,骨髓出现大量铁颗粒,形成环形铁粒幼细胞。遗传性SA主要为X连锁性铁幼粒细胞贫血(XLSA)[1]。XLSA发病可能是由于红细胞系统特异性δ氨基γ酮戊酸合成酶2(ALAS2)基因突变所致[2]。我们最近发现一个3代铁粒幼细胞贫血家系,4兄妹中有2个兄弟发病。用与ALAS2距离小于1.7Mb的微卫星做连锁分析以了解该家系的遗传方式;克隆患者及正常人ALAS2基因的cDNA全编码区并测序。发现患者ALAS2基因存在一种新的突变,第5外显子A523G突变,导致苏氨酸变为丙氨酸。
对象和方法
, 百拇医药 1 病例
1.1 家系:先证者兄弟2人及其家系3代共9个成员,其中第1代1人,第2代6人,即先证者兄弟2人及其配偶1人,先证者之弟、妹2人及其配偶1人;第3代2人,其中之一为患者之子。均进行血常规、血清铁,铁蛋白、总铁结合力测定,血涂片检查,第1代母亲无贫血但红细胞呈双形性。第2代中患者兄弟2人均以糖尿病为首发症状,血清铁高,均在20岁左右出现低色素、小细胞贫血,骨髓可见大量环形铁粒幼细胞,血清铁明显增高。第2代另外4名成员和第3代2名男性后代正常。
1.2 先证者1:男,47岁。因“发现贫血27年,多饮、多尿、体重下降3个月”入院。27年前偶然发现贫血,按“缺铁性贫血”治疗数年无效。20年前始面色青灰。3个月前出现多饮、多尿、体重下降。空腹血糖3.9g/L。查体:全身皮肤呈青灰色,肝肋下2cm,质硬,边钝,表面光滑,压痛(+),脾肋缘下4~5cm,质硬,边钝,表面光滑,压痛(+),双下肢轻度可凹性水肿。血常规:Hb 59g/L,RBC 2.55×1012/L, WBC 2.7×109/L,BPC 236×109/L,网织0.016;骨髓象:骨髓增生明显活跃,红系52.5%,中幼红细胞增高,成熟红细胞明显大小不等,畸型红细胞多见,部分红细胞中心浅染区扩大,粒、红细胞数量比为0.7∶1,全片见38个巨核细胞;细胞外铁(++),细胞内铁阳性率0.63,其中Ⅰ型0.15,Ⅱ型0.02, Ⅲ型0.05,Ⅳ型0.23,环型0.18。铁蛋白>500μg/L,血清铁 48μmol/L,总铁结合力47μmol/L。馒头餐:血糖0,1,2,3h分别为:20.38, 24.71,28.50, 26.02mmol/L;胰岛素0,1,2,3h分别为:4.16,4.56,4.74,5.00mIU/L。T3 0.47mg/L (正常0.69~2.15mg/L),T4 39.8 mg/L (正常42.3~112.5mg/L),促甲状腺素(TSH)<0.03 mg/L (正常0.75~3.53mg/L)。尿17羟12.6mg/24h;尿17酮16.2mg/24h。促肾上腺皮质素(ACTH)114.44ng/L(正常6.0~56.7ng/L)。B超:肝大,肝实质回声致密,门脉主干宽1.4cm,脾大,肋缘下平脐,下腹盆腔可见少量积液。超声心动图:各房室大小正常,二尖瓣前后叶稍增厚,轻度关闭不全,瓣膜活动正常,室壁不厚,运动正常。Rous试验四次(-);蔗糖水试验(-);Ham试验(-);CD59阳性率100%;Coombs试验(-)。血红蛋白电泳两次(-)。尿卟胆原(-),G6PD酶活性正常。入院后予诺和灵降血糖,但多次发生低血糖发作,曾心悸,心电图示快速房颤,10min后自行缓解。诊断为铁粒幼细胞贫血,继发性血色病,继发性糖尿病。2个月后因顽固性心律失常心衰死亡。
, 百拇医药
1.3 先证者2,男,38岁。因“发现贫血20 余年,发现血糖高4个月”入院。20余年前查体发现 “缺铁性贫血”,因口渴,多饮,尿量增多,血糖高入院。查体:面色苍白,稍铁青,肝、脾触诊不满意,双踝部可凹性水肿。实验室检查:馒头餐试验血糖呈典型糖尿病曲线,铁蛋白>500μg/L,血清铁>90μmol/L,总铁结合力32.1μmol/L,网织红细胞不高,Coombs试验(-),Rous试验(-)。骨髓象:骨髓增生活跃,粒系0.31,粒、红细胞数量比为0.6∶1;红系比例0.50,中幼红细胞0.32,晚幼红细胞0.17,有核红细胞胞体小,胞浆少,成熟红细胞大小不等明显,中心浅染区扩大,畸型红细胞及大红细胞多见。全片见1个巨核细胞。细胞外铁(-),细胞内铁阳性率0.90,其中Ⅰ型0.06,Ⅱ型0.18,Ⅲ型0.22,Ⅳ型0.12,环型0.32。骨髓活检:符合铁粒幼细胞贫血。MRI提示肝、脾、胰脏信号异常,脾大,考虑铁沉积。血红蛋白电泳(两次):正常。T3 0.59 mg/L,T4 35.6 mg/L, TSH 2.81 mg/L。尿3-甲氧-4羟苦杰仁酸(VMA) <5mg/24h;尿糖定量52.5g/24h;尿17羟14.0mg/24h, 尿17酮10.5 mg/24 h。超声心动图:左房、室扩大并二尖瓣关闭不全; 主动脉瓣轻度关闭不全,左室收缩功能降低。诊断为铁粒幼细胞贫血,继发性血色病、糖尿病。
, http://www.100md.com
2 标本 取外周血细胞用高盐沉淀法提取DNA做基因分析;10名正常人外周血DNA和骨髓作为微卫星多态性正常对照;从患者骨髓细胞和3个月龄人工流产正常胎儿肝脏提取总mRNA,poly(dT)引物制备cDNA,以克隆ALAS2的cDNA。
3 PCR引物设计与合成 从国际互联网www.genethon.fr基因信息库中查得3对微卫星引物,送赛百盛公司合成。其中包括染色体Xp11.21上与ALAS 2距离小于1.7Mb的微卫星DXS 991和DXS 1199以及位于Xp22.13的DXS 1226。ALAS2 cDNA克隆的引物参照Genebank x56352的序列设计,有义链: 5′-ACTAGGATCCGCAGCCATGCTGCTACAGTGC-3′,反义链:5′-CTAGAATTCAGGCATAGGTGGTGACATAC-3′。
4 PCR扩增微卫星 25μl反应缓冲液中含有:模板DNA 0.25ng,Mg离子2mmol/L,4×dNTP 各50μmol/L,引物各10pmol。 Taq聚合酶0.5U。PCR反应程序:94℃ 5min变性后,进行以下30个循环:93℃ 30s,56℃ 30s,72℃ 1min。之后72℃延伸10min。实验重复2次以上。取PCR扩增反应液4~8μl加入1~2倍含体积分数为95%甲酰胺的溴酚蓝上样缓冲液,在含7mol/L尿素的60g/L的聚丙烯酰胺凝胶上,300 V进行电泳,银染显色。
, 百拇医药
5 从先证者2的骨髓和胎肝细胞中克隆ALAS2 cDNA的全部编码区 用淋巴细胞分离液分离骨髓有核细胞。骨髓有核细胞和胎肝细胞总RNA提取采用Gibco/BRL公司的TRIZOL试剂盒,按照说明提取RNA。 逆转录用该公司的Superscript cDNA第一链合成试剂盒,以poly(dT)为引物,逆转录反应的总体系为20μl。PCR反应体系25μl,含逆转录反应产物cDNA 1μl(作为PCR反应模板),有义链和反义链引物各25pmol,94℃预变性5min; 循环温度为94℃变性1min,54℃退火1min,72℃延伸90s,共30个循环; 循环结束后继续于72℃延伸7min。反应产物用聚丙烯酰胺凝胶电泳,银染法鉴定。
6 先证者2和胎儿ALAS2基因片段进行克隆和限制性酶谱分析 将25μl PCR产物经酚提取,乙醇沉淀后溶于5μl TE中,用BamHⅠ和EcoRⅠ酶消化1h,再用低熔点琼脂糖凝胶纯化。定向克隆于质粒pcDNA3 BamHⅠ和EcoRⅠ位点中。选择有克隆产物的质粒,BamHⅠ+EcoRⅠ和BstⅠ消化后,根据片段长度做鉴定。
, 百拇医药
7 测序分析 用ABI测序仪分3段正反向测序,分析全部1800bp左右的序列。中段测序引物5′-CCCAACATTCTGAG-3′。
结果
1 正常人DNA微卫星DXS 991和DXS 1199以及DXS 1226片段多态性 从10名正常人采集的外周血提取DNA,然后进行了3个微卫星位点的分析,结果不同个体的微卫星DXS 991和DXS 1199以及DXS 1226片段长短不同,呈多态性,可用作单体型分析。
2 先证者家系遗传连锁分析结果 2例先证者DXS 991和DXS 1199的多态性相联锁,也与母亲DXS 991和DXS 1199两等位基因之一相一致。提示患病兄弟从母系获得同样的致病性ALAS2基因。
3 用RT-PCR克隆获得先证者和正常胎肝造血细胞ALAS2 cDNA全部编码区 患者cDNA克隆的pcDNA经限制性内切酶BamHⅠ和EcoRⅠ消化电泳后证实为1700bp序列;胎肝则获得1700bp和1600bp两条片段(图1)。
, 百拇医药
1,4:从胎肝获得1700和1600bp 2条ALAS2基因片段;
2,3:先证者获得1700bp 1条片段;M为分子量标志
图1 RT-PCR克隆ALAS2基因片段
4 克隆片段的限制性酶谱分析 先证者和胎儿的ALAS2/pcDNA3质粒均可被PstⅠ消化获得4500或4600,1385,595及540 bp 4条新的较小片段。与报道的ALAS2 cDNA相符(图2)。
M:分子量标准λ DNA/EcoR Ⅰ+Hind Ⅲ; 1:病例2;
2:正常胎肝的1700bp片段; 3:正常胎肝的1600bp的片段
, http://www.100md.com
图2 ALAS2/pcDNA3克隆的PstⅠ酶切片段
5 经测序,从胎肝细胞获得的两ALAS2克隆,大片段与正常的ALAS2 cDNA序列(x60364)相同,小片段缺失第4外显子111 bp序列。
6 先证者与胎儿ALAS2序列对比分析 比较先证者和胎肝ALAS2发现,先证者第5外显子A523G突变(图3),导致苏氨酸变为丙氨酸;即ALAS2蛋白的T161A突变;第3外显子T372C突变,导致亮氨酸由脯氨酸替代,即ALAS2蛋白的L107P的突变。后者在ALAS2可剪拼区的第4外显子内。
图3 测序发现先证者ALAS2基因第5外显子A523G突变
讨论
, http://www.100md.com SA是一组铁利用障碍所致的贫血性疾病。遗传性SA主要为X连锁SA(XLAS)。最近发现XLSA发病是由于红细胞线粒体中的δ氨基γ酮戊酸合成酶2(ALAS2)基因突变所致[2]。这些突变发生在第5~11外显子中,至1999年已有15种[3-5]。我们最近在北京地区发现一个XLSA家系,4兄妹中有2个兄弟发病,先用与ALAS2距离小于1.7Mb的微卫星DXS 991和DXS 1199以及距离较远、位于Xp22.13的DXS1226做连锁分析,两兄弟都从母系获得同样的DXS 991和DXS 1199基因。而患者距离较远的DXS 1226不连锁。表明该家系两患者兄弟都从母系获得同一ALAS2等位基因。健康兄、妹2人则获得另一条ALAS2等位基因。随后克隆患者和正常人ALAS2 cDNA编码区,经限制性酶谱和测序发现,患者第5外显子A523G突变,导致苏氨酸变为丙氨酸;这是国内外尚未报告过的一种新ALAS2基因突变, 也是首次在国内发现导致铁粒幼细胞贫血的基因突变。我们同时发现患者第4外显子T372C突变,可导致亮氨酸由脯氨酸替代,但这个突变位点在ALAS2 cDNA的可剪拼区内。Canboy等分析ALAS2基因发现其存在两种不同的异构体,一种异构体比另一种少37个氨基酸、111个碱基,缺失部分为ALAS2基因的第4外显子。他们认为这种缺失片段没有明显的功能差异。因此该突变可能不是该家族患病的致病基因位点,其意义有待进一步分析。从临床表现结合文献报道分析,这种X连锁的疾病有如下特征:往往在20岁左右发现贫血和症状;由于不适当地输血和补铁造成铁负荷过重,中年易出现继发性血色病,可并发糖尿病、肝硬化或者心肌病等;血清铁、铁蛋白明显增高;骨髓出现大量环形铁幼粒细胞。一些女性携带者可有脾肿大和少数红细胞异常,但是没有贫血。
, 百拇医药
基金项目:国家自然科学基金资助项目(3957321)和卫生部科研基金资助项目(98-2-2220)
参 考 文 献
1,Bottomley SS, Wise PD, Wassson EG, et al. X-linked sideroblastic anemia: update of molecular detects and mechanism of the associated iron overload. Acta Haematologica, 1997, 98:103.
2,Conboy JH, Cox TC, Bottemley S, et al. Human erythriod 5-aminolevulinate synthese gene structure and specific differences in alternative RNA splicing. J Bio Chem,1992, 267:18753-18758.
, 百拇医药
3,Cotter PD, May A, Li L, et al. Four new mutations in the erythroid-specific 5-aminolevulinate synthase (ALAS2) gene causing X-linked sideroblastic anemia: increased pyridoxine responsiveness after removal of iron overload by phlebotomy and coinheritance of hereditary hemochromatosis. Blood, 1999, 93: 1757-1769.
4,Cox TC, Bottomley SS, Wiley JS. et al. X-linked pyridoxine-responsive sideroblastic anemia due to a thr388-to-ser substitution in erythroid 5-aminolevulinate synthase. New Eng J Med, 1994. 330: 675-679.
5,Prades E, Chambon C, Dailey TA,et al. A new mutation of the ALAS2 gene in a large family with X-linked sideroblastic anemia. Hum Genet, 1995, 95: 424-428.
(收稿日期:1999-09-13), 百拇医药