脊肌萎缩症的产前基因诊断
作者:袁丽芳 刘天慈 马素参 周文敏 杨涛 吴沪生 孙念怙 赵时敏
单位:袁丽芳 马素参 杨涛 100005北京,中国医学科学院基础医学研究所 中国协和科大学基础医学院医学遗传室;刘天慈 周文敏 吴沪生 首都医科大学附属儿童医院;孙念怙 赵时敏 北京协和医院
关键词:
脊肌萎缩症的产前基因诊断 脊肌萎缩症(spinal muscular atrophy,SMA)是较常见的常染色体隐性遗传病。在白种人中发生率为1/6000[1],我国尚无统计,但不少见。根据病情轻重及病程,本病可分3型[2]:Ⅰ型又称重型,急性型、婴儿型,或Werdnig-Hoffman disease;Ⅱ型又称中间型;Ⅲ型又称Kuglberg-Walender disease。由于本病目前尚无治疗方法,再现率高,许多家庭要求产前诊断。为建立产前诊断方法,我们对46个SMA患儿作运动神经原存活基因(survival motor neuron,SMN)基因缺失的检测,对36个SMA家系应用3个与SMA基因紧密连锁的CA重复序列位点JK53CAl/2、5DS637及CATT1进行家系连锁分析。
, 百拇医药
1 材料与方法
1.1 材料
1.1.1 检测对象 46例患儿均符合SMA诊断标准[2]:(1)临床表现符合脊髓前角运动神经原病变:肌力、肌张力减弱,腱反射减弱或消失,肌萎缩,纤颤;(2)血清CPK正常或稍高;(3)肌电图:前角运动神经原损害;(4)肌活检符合SMA病变;(5)家族史符合常染色体隐性遗传,有家族史者免活检。Ⅰ型于6个月内发病,不能独坐,2岁前死亡;Ⅱ型于1岁内发病,能独坐,但不能行走;Ⅲ型于3岁后发病,能行走,但肌无力。
1.1.2 DNA来源 46例患儿来自北京市儿童医院及北京协和医院儿科遗传门诊。Ⅰ型18例,Ⅱ型28例。10名正常人作为对照。取患儿及其父母(家系核心成员)以及正常人血用盐析法提取DNA,产前诊断取绒毛或羊水细胞DNA用苯酚、氯仿法提取。
1.2 方法
, http://www.100md.com
1.2.1 SMA基因缺失的检测 对46例SMA患儿用聚合酶链反应-单链构象多态性(polymerase chain reaction-single strand conformation polymorphism,PCR-SSCP)分析作端粒侧(Tel)外显子7的检测。引物序列、SSCP分析方法见文献[3]。
1.2.2 PCR扩增SMA基因旁侧CA重复序列作家系连锁分析 (1)36个SMA家系成员的基因组DNA用3对SMA旁侧CA重复序列位点:JK53CA1/2,5DS637,CATT1进行PCR扩增,这三个位点均位于5q13,其特性、序列及扩增条件见文献[4]。(2)PCR产物分别行6%~8%变性聚丙烯酰胺电泳。电泳条件:1×TBE,电压580V,电流30mA,2~4小时。(3)银染 连锁分析方法见文献[4]。
1.3 7例SMA产前基因诊断 有两对夫妇生过1个SMA Ⅰ型患儿,已病故。于孕期9~10周取绒毛、或16周后取羊水提取DNA作PCR-SSCP分析,无先证者样品的家系仅测胎儿是否有SMA基因缺失,其他5个核心家系均作基因缺失检测及家系连锁分析。5例于孕早期9~10周取绒毛,1例取羊水细胞分析,另1例同时作绒毛及羊水细胞分析。
, http://www.100md.com
2 结果
2.1 基因缺失检测 40例有SMN基因端粒侧外显子7的纯合性缺失,其中一例的母亲、另一例的父亲也有同样的纯缺失。6例未见基因缺失,缺失率为87%。
2.2 核心家系作连锁分析结果 JK53CA1/2位点的可诊断率为25/36(70%),5DS637的可诊断率为30/36(84%),CATTⅠ为15/16(94%)。
2.3 7例产前诊断结果 3例胎儿PCR-SSCP分析结果为父母正常,胎儿有SMN Tel外显子7的纯合性缺失(图1),其中1例同时以5DS637作家系连锁分析,证明胎儿患病(图2),终止妊娠。其他4例PCR-SSCP分析未见SMN Tel外显子7基因纯合性缺失,这四个家系都作了家系连锁分析,3例于5DS637位点获得信息,1例根据5DS637及JK53CA1/2两个位点作出判断。连锁分析结果说明胎儿为杂合子或正常纯合子。产前诊断结果判断为正常的胎儿均已出生,年龄分别为2个月、8个月、1岁2个月及1岁5个月,精神运动发育正常。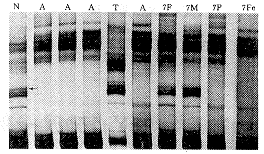
, http://www.100md.com
图1 应用PCR-SSCP检测SMATel外显子7 进行SMA产前诊断(例7) 箭头显示胎儿SMN外显子7缺失.N:正常对照;A:患儿;T:SMN Tel外显子7标准;F:父亲;M:母亲;P:先证者;Fe:胎儿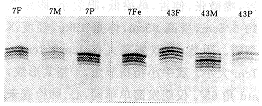
图2 PCR及8%变性胶电泳 用5DS637位点作家系7连锁分析,进行SMA产前基因诊断。结果显示:羊水细胞的带型与患儿相同,与母亲不同,说明胎儿患病;F:父亲;M:母亲;P:先证者;Fe:胎儿(羊水细胞DNA)
3 讨论
1990年Brzustowicz[5]及Melki[6]等将三型SMA定位于5qll-5q13。此后不少学者在此区域找到许多与SMA基因紧密连锁的多态位点,为家系连锁分析提供了良好条件。1995年Lefebvre[7]及Roy[8]两组实验室分别克隆分离出2个SMA候选基因,各自命名为运动神经原存活基因及神经原凋亡抑制蛋白基因(neuronal-apoptosis inhibitory protein,NAIP),二者都能满足基因条件,均位于5q13。并各有2个拷贝,一个位于着丝粒侧,另一个位于端粒侧,认为端粒侧具有功能。Lefebvre等[7]对229例SMA患者作PCR-SSCP分析,发现其中98%有SMN Tel外显子7的纯合性缺失。其他学者报道的缺失率为84%~100%。我们应用PCR-SSCP对46例SMA患儿检测SMN Tel外显子7是否缺失,并用与SMA基因紧密连锁的3个CA重复序列位点:JK53CA1/2、5DS637及CATT1,对36个SMA家系进行家系连锁分析,在此基础上作了7例SMA的产前基因诊断。结果说明,应用SMA基因旁侧CA重复序列作家系连锁分析联合SMN外显子7缺失检测,可有效地进行SMA的基因诊断及产前诊断。方法简便,可靠,快速,适用于临床。此外,由于SMA患儿的SMN Tel外显子7纯合性缺失率高达87%以上,因此可用于临床诊断SMA,取代肌活检,且特异性更高。至于1个SMA患儿的母亲也发现有SMN Tel外显子7的纯合缺失,经一些作者分析认为,他们虽有SMN Tel外显子7的缺失,但SMN着丝粒侧(Cen)外显子7的拷贝数增加,SMN Cen外显子7作用虽弱,但拷贝数增加后可以完全代偿SMN Tel外显子7的功能[9],因而不出现症状。产前诊断必须严格、谨慎,尽可能于孕早期取绒毛,细心挑取绒毛至关重要。如果胎儿的基因型与母亲相同,必须选作另一条染色体上的多态位点进行分析,以排除样品为母体细胞污染的可能。
, 百拇医药
志谢 本课题承北京市协和医院郭玉璞教授、任海涛同志协助作病理学检查,Dr.Lefebvre惠赠SMNexon7标准,特致谢意
本项目受卫生部科学研究基金(94-1-012)资助
参考文献
1 Lewin B.Genes for SMA:Multum in parvo.Cell,1995,80(1)∶1-5.
2 Cobben JM,Visser M de,Scheffer H,et al.Confirmation of clinical diagnosis on requests for prenatal prediction of SMA type Ⅰ. J Neurol Neurosurg Psychiatry, 1993,56(3)∶319-321.
, 百拇医药
3 杨涛,袁丽芳,刘天慈,等.脊肌萎缩症基因缺失的初步研究.中华医学遗传学杂志,1998,15(2)∶95-97.
4 袁丽芳,刘天慈,杨涛,等.儿童期脊肌萎缩症的基因诊断及产前基因诊断.中华儿科杂志,1997,35(12)∶631-634.
5 Burzustowicz LM,Lehner T,Castilla LH,et al.Genetic mapping of chronic childhood-onset spinal muscular atrophy to chromosome 5q11.2-q13.Nature,1990,344(6266)∶540-541.
6 Melki J,Sheth P,Abdelhak S,et al.Mapping of acute (type Ⅰ) spinal muscular atrophy to chromosome 5q12-q14.Lancet,1990,336(8710)∶171-173.
, 百拇医药
7 Lefebvre S,Burgten L,Reboullet S,et al.Identification and characterization of a spinal muscular atrophy-determining gene.Cell,1995,80(1)∶155-165.
8 Roy N,Mahadevan MS,McLean M,et al.The gene for neuronal apoptosis inhibitory protein is partially deleted in individuals with spinal muscular atrophy.Cell,1995,80(1)∶167-168.
9 Campbell L,Potter A,Ignatius J,et al.Genomic variation and gene onversion in spinal muscular atrophy:implication for disease processs and clinical phenotype.Am J Hum Genet,1997,61(4)∶40-50.
(收稿:1999-02-25 修回:1999-04-15), http://www.100md.com
单位:袁丽芳 马素参 杨涛 100005北京,中国医学科学院基础医学研究所 中国协和科大学基础医学院医学遗传室;刘天慈 周文敏 吴沪生 首都医科大学附属儿童医院;孙念怙 赵时敏 北京协和医院
关键词:
脊肌萎缩症的产前基因诊断 脊肌萎缩症(spinal muscular atrophy,SMA)是较常见的常染色体隐性遗传病。在白种人中发生率为1/6000[1],我国尚无统计,但不少见。根据病情轻重及病程,本病可分3型[2]:Ⅰ型又称重型,急性型、婴儿型,或Werdnig-Hoffman disease;Ⅱ型又称中间型;Ⅲ型又称Kuglberg-Walender disease。由于本病目前尚无治疗方法,再现率高,许多家庭要求产前诊断。为建立产前诊断方法,我们对46个SMA患儿作运动神经原存活基因(survival motor neuron,SMN)基因缺失的检测,对36个SMA家系应用3个与SMA基因紧密连锁的CA重复序列位点JK53CAl/2、5DS637及CATT1进行家系连锁分析。
, 百拇医药
1 材料与方法
1.1 材料
1.1.1 检测对象 46例患儿均符合SMA诊断标准[2]:(1)临床表现符合脊髓前角运动神经原病变:肌力、肌张力减弱,腱反射减弱或消失,肌萎缩,纤颤;(2)血清CPK正常或稍高;(3)肌电图:前角运动神经原损害;(4)肌活检符合SMA病变;(5)家族史符合常染色体隐性遗传,有家族史者免活检。Ⅰ型于6个月内发病,不能独坐,2岁前死亡;Ⅱ型于1岁内发病,能独坐,但不能行走;Ⅲ型于3岁后发病,能行走,但肌无力。
1.1.2 DNA来源 46例患儿来自北京市儿童医院及北京协和医院儿科遗传门诊。Ⅰ型18例,Ⅱ型28例。10名正常人作为对照。取患儿及其父母(家系核心成员)以及正常人血用盐析法提取DNA,产前诊断取绒毛或羊水细胞DNA用苯酚、氯仿法提取。
1.2 方法
, http://www.100md.com
1.2.1 SMA基因缺失的检测 对46例SMA患儿用聚合酶链反应-单链构象多态性(polymerase chain reaction-single strand conformation polymorphism,PCR-SSCP)分析作端粒侧(Tel)外显子7的检测。引物序列、SSCP分析方法见文献[3]。
1.2.2 PCR扩增SMA基因旁侧CA重复序列作家系连锁分析 (1)36个SMA家系成员的基因组DNA用3对SMA旁侧CA重复序列位点:JK53CA1/2,5DS637,CATT1进行PCR扩增,这三个位点均位于5q13,其特性、序列及扩增条件见文献[4]。(2)PCR产物分别行6%~8%变性聚丙烯酰胺电泳。电泳条件:1×TBE,电压580V,电流30mA,2~4小时。(3)银染 连锁分析方法见文献[4]。
1.3 7例SMA产前基因诊断 有两对夫妇生过1个SMA Ⅰ型患儿,已病故。于孕期9~10周取绒毛、或16周后取羊水提取DNA作PCR-SSCP分析,无先证者样品的家系仅测胎儿是否有SMA基因缺失,其他5个核心家系均作基因缺失检测及家系连锁分析。5例于孕早期9~10周取绒毛,1例取羊水细胞分析,另1例同时作绒毛及羊水细胞分析。
, http://www.100md.com
2 结果
2.1 基因缺失检测 40例有SMN基因端粒侧外显子7的纯合性缺失,其中一例的母亲、另一例的父亲也有同样的纯缺失。6例未见基因缺失,缺失率为87%。
2.2 核心家系作连锁分析结果 JK53CA1/2位点的可诊断率为25/36(70%),5DS637的可诊断率为30/36(84%),CATTⅠ为15/16(94%)。
2.3 7例产前诊断结果 3例胎儿PCR-SSCP分析结果为父母正常,胎儿有SMN Tel外显子7的纯合性缺失(图1),其中1例同时以5DS637作家系连锁分析,证明胎儿患病(图2),终止妊娠。其他4例PCR-SSCP分析未见SMN Tel外显子7基因纯合性缺失,这四个家系都作了家系连锁分析,3例于5DS637位点获得信息,1例根据5DS637及JK53CA1/2两个位点作出判断。连锁分析结果说明胎儿为杂合子或正常纯合子。产前诊断结果判断为正常的胎儿均已出生,年龄分别为2个月、8个月、1岁2个月及1岁5个月,精神运动发育正常。
, http://www.100md.com
图1 应用PCR-SSCP检测SMATel外显子7 进行SMA产前诊断(例7) 箭头显示胎儿SMN外显子7缺失.N:正常对照;A:患儿;T:SMN Tel外显子7标准;F:父亲;M:母亲;P:先证者;Fe:胎儿
图2 PCR及8%变性胶电泳 用5DS637位点作家系7连锁分析,进行SMA产前基因诊断。结果显示:羊水细胞的带型与患儿相同,与母亲不同,说明胎儿患病;F:父亲;M:母亲;P:先证者;Fe:胎儿(羊水细胞DNA)
3 讨论
1990年Brzustowicz[5]及Melki[6]等将三型SMA定位于5qll-5q13。此后不少学者在此区域找到许多与SMA基因紧密连锁的多态位点,为家系连锁分析提供了良好条件。1995年Lefebvre[7]及Roy[8]两组实验室分别克隆分离出2个SMA候选基因,各自命名为运动神经原存活基因及神经原凋亡抑制蛋白基因(neuronal-apoptosis inhibitory protein,NAIP),二者都能满足基因条件,均位于5q13。并各有2个拷贝,一个位于着丝粒侧,另一个位于端粒侧,认为端粒侧具有功能。Lefebvre等[7]对229例SMA患者作PCR-SSCP分析,发现其中98%有SMN Tel外显子7的纯合性缺失。其他学者报道的缺失率为84%~100%。我们应用PCR-SSCP对46例SMA患儿检测SMN Tel外显子7是否缺失,并用与SMA基因紧密连锁的3个CA重复序列位点:JK53CA1/2、5DS637及CATT1,对36个SMA家系进行家系连锁分析,在此基础上作了7例SMA的产前基因诊断。结果说明,应用SMA基因旁侧CA重复序列作家系连锁分析联合SMN外显子7缺失检测,可有效地进行SMA的基因诊断及产前诊断。方法简便,可靠,快速,适用于临床。此外,由于SMA患儿的SMN Tel外显子7纯合性缺失率高达87%以上,因此可用于临床诊断SMA,取代肌活检,且特异性更高。至于1个SMA患儿的母亲也发现有SMN Tel外显子7的纯合缺失,经一些作者分析认为,他们虽有SMN Tel外显子7的缺失,但SMN着丝粒侧(Cen)外显子7的拷贝数增加,SMN Cen外显子7作用虽弱,但拷贝数增加后可以完全代偿SMN Tel外显子7的功能[9],因而不出现症状。产前诊断必须严格、谨慎,尽可能于孕早期取绒毛,细心挑取绒毛至关重要。如果胎儿的基因型与母亲相同,必须选作另一条染色体上的多态位点进行分析,以排除样品为母体细胞污染的可能。
, 百拇医药
志谢 本课题承北京市协和医院郭玉璞教授、任海涛同志协助作病理学检查,Dr.Lefebvre惠赠SMNexon7标准,特致谢意
本项目受卫生部科学研究基金(94-1-012)资助
参考文献
1 Lewin B.Genes for SMA:Multum in parvo.Cell,1995,80(1)∶1-5.
2 Cobben JM,Visser M de,Scheffer H,et al.Confirmation of clinical diagnosis on requests for prenatal prediction of SMA type Ⅰ. J Neurol Neurosurg Psychiatry, 1993,56(3)∶319-321.
, 百拇医药
3 杨涛,袁丽芳,刘天慈,等.脊肌萎缩症基因缺失的初步研究.中华医学遗传学杂志,1998,15(2)∶95-97.
4 袁丽芳,刘天慈,杨涛,等.儿童期脊肌萎缩症的基因诊断及产前基因诊断.中华儿科杂志,1997,35(12)∶631-634.
5 Burzustowicz LM,Lehner T,Castilla LH,et al.Genetic mapping of chronic childhood-onset spinal muscular atrophy to chromosome 5q11.2-q13.Nature,1990,344(6266)∶540-541.
6 Melki J,Sheth P,Abdelhak S,et al.Mapping of acute (type Ⅰ) spinal muscular atrophy to chromosome 5q12-q14.Lancet,1990,336(8710)∶171-173.
, 百拇医药
7 Lefebvre S,Burgten L,Reboullet S,et al.Identification and characterization of a spinal muscular atrophy-determining gene.Cell,1995,80(1)∶155-165.
8 Roy N,Mahadevan MS,McLean M,et al.The gene for neuronal apoptosis inhibitory protein is partially deleted in individuals with spinal muscular atrophy.Cell,1995,80(1)∶167-168.
9 Campbell L,Potter A,Ignatius J,et al.Genomic variation and gene onversion in spinal muscular atrophy:implication for disease processs and clinical phenotype.Am J Hum Genet,1997,61(4)∶40-50.
(收稿:1999-02-25 修回:1999-04-15), http://www.100md.com