新红霉素衍生物的结构修饰与抗菌活性研究进展
作者:许先栋
单位:(中国医学科学院 中国协和医科大学 医药生物技术研究所,北京 100050)
关键词:红霉素衍生物;构效关系
新红霉素衍生物的结构修饰与抗菌活性研究进展 摘要 综述了近年来红霉素衍生物的结构修饰与抗菌活性关系研究的最新进展,结果显示,该红霉素类衍生物新药品种具有组织和血药浓度高,半衰期长和抗耐药菌等药代动力学新特性。
PROGRESS IN STRUCTURE-ACTIVITY RELATIONSHIP STUDY OF
ERYTHROMYCIN DERIVATIVES
Xu Xiandong
, 百拇医药 (Institute of Medicinal Biotechnology,Peking Union Medical College,Chinese Academy of Medical Sciences,Beijing 100050)
ABSTRACT Recent progress in structure modification of erythromycin derivatives,their antibacterial activities,and pharmacokinetics were reviewed in this paper.
KEY WORDS Erythromycin derivatives;Structure-activity relationship
近20年来,根据红霉素酸缩酮化失效原理,世界药学工作者广泛深入地开展了对红霉素的结构修饰与抗菌活性研究,获得了一系列具有药代动力学新特性的红霉素类大环内酯新药品种,如罗红霉素(roxithromycin)、阿齐霉素(azithromycin)、克拉霉素(clarithromycin)、地红霉素(dirithromycin)、氟红霉素(flurithromycin)等红霉素衍生物(俗称第二代红霉素)。解决了红霉素因酸缩酮化形成8,9-脱水红霉素-6,9-半缩酮而失效,从而提高了血药浓度,延长了半衰期,改进了组织移行性,减少了用药量和副作用,增强了疗效。但这些新红霉素衍生物不仅抗菌谱、临床适应证与已知大环内酯抗生素相同,而且仍具有诱导耐药性。因此改善其耐药性,扩展其抗菌谱,增强其抗菌活性依然是该类抗生素研究的重要方向。新近Rorssel Uclaf和Abbott实验室等报道,红霉素内酯环C-3位脱cladinose sugar后转变为C-3位酮基红霉素,其衍生物的作用特点是,克服了十四元大环内酯所共有的耐药性,对大环内酯耐药菌具有较好的抗菌活性,如对耐大环内酯的肺炎球菌、金葡菌有优异的抗菌活性,对抗肠球菌活性亦增强以及对嗜血杆菌属亦有活性。
, 百拇医药
现就红霉素A分子结构中可供修饰的基团进行的结构修饰特点和抗菌活性关系作一简要综述。
1 内酯环6,11,12,和糖上2′,4″位羟基的修饰
基于内酯环上羟基的醚化,可阻止其酸性条件下分子内缩酮化形成螺缩酮,从而使其衍生物具有对酸稳定,可口服吸收快,血药浓度高而持久的特点。1981年日本大正制药公司从红霉素A出发,经肟化、醚化和硅烷化、甲基化和还原制得克拉霉素。本品对需氧菌、支原体、脲原体和衣原体等有效,体内活性比红霉素强2~4倍。又如C-6,C-11位羟基同时烷基化制得6,11-二氧烷基红霉素,其代表衍生物6,11-二甲氧基红霉素口服比红霉素A抗菌作用强4~9倍,且血药浓度高而持久。
关于内酯环上11,12位羟基同时修饰,Abbott实验室和Slawinski等曾制得一系列红霉素和克拉霉素的11,12位环状氨基甲酸酯,其代表衍生物A-66321的体外活性表明,增强了对结构耐药菌如化脓性链球菌的抗菌活性。又如Brain等在红霉素11,12位环状氨基甲酸酯的9位酮基进行肟化,同时辅以其他位置的取代制得一系列衍生物。其体内、外活性表明,对多种G+和G-菌有不同程度的抗菌活性,且未见明显毒副作用。
, http://www.100md.com
关于糖上2′,4″位羟基进行酰化制得的多种酯类衍生物如2′位乙酰酯、丙酰酯、碳酸乙酯、乙基丁二酰酯和4″-氧-乙酰胺基-6-氧甲基红霉素均已被确认为有效新型大环内酯类抗生素。
2 C-9位酮基的修饰
红霉素9位酮基最简便的修饰是变为肟基,从而阻止其分子内酸缩酮化。1972年USpatent 368326报道,利用RONH2。HX与红霉素A或B作用制得其相应肟化衍生物,并显示一定的抗菌活性。之后Jean-claude等继而对红霉素肟羟基进行醚化制得60多种醚化衍生物,经构效关系研究证明,该醚肟衍生物无论在体内、体外均具有强于红霉素A的抗菌活性,其中14种衍生物的药代动力学性质有明显改善。此外,经对肟基双键的顺反异构体的抗菌活性对照试验,发现所有顺式异构体抗菌活性较低。之后Roussel Uclaf公司通过优选法得到RU28965,即罗红霉素,并从红霉素A出发经肟化再与甲氧基乙氧基甲基氯缩合制得其产品,其组织中分布广,尤其在肺组织中浓度较高。
, 百拇医药
C-9位酮基亦可转变为胺基而制得红霉素胺(erythromycylamine),它对G+菌作用较弱,但对C-菌作用有所增强。由于其口服吸收差,后由德国Karl Thomae公司经由红霉素A胺化、重氮化、还原制得红霉素胺,再与甲氧基乙氧基乙醛缩合制得地红霉素。由于其分子结构中的C-9位胺基和11位羟基与侧链醛缩合形成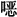 嗪环,阻止了6,9和9,12位螺缩酮的形成而对酸和酶稳定,半衰期长达20~50 h,增强了对G+菌、幽门螺旋菌和衣原体、军团菌活性,并具有一定的免疫调节作用和良好的药代动力学性质。
嗪环,阻止了6,9和9,12位螺缩酮的形成而对酸和酶稳定,半衰期长达20~50 h,增强了对G+菌、幽门螺旋菌和衣原体、军团菌活性,并具有一定的免疫调节作用和良好的药代动力学性质。
此外,Plive和Pfizer公司实验室曾将红霉素9位酮基肟化后进行Beckmann重排和N上甲基化反应,得具有十五元含氮大环内酯类抗生素阿奇霉素,本品对酸稳定,组织浓度大于血药浓度,半衰期长,其抗菌特点是对需氧和厌氧G-菌及细胞内病原菌、支原体和衣原体有较强的抗菌作用。另于1988年EP 0307128专利报道,阿齐霉素和4″位差向异构体和4″-去氧-4″-氨基类似物的抗原虫活性。Gabrijela等还报道了阿奇霉素-氧-甲基衍生物的制备,测定了它们的抗菌活性,推断了它们的构效关系。
, 百拇医药
3 C-8位质子的修饰
由于C-8位质子可用负电性强的离子或基团取代,80年代意大利Pierrel公司开发了8(S)-8-氟红霉素(flurithromycin),是将红霉素A在乙酸作用下转变为8,9-烯醇式半缩醛,再与FClO3或CF3OF和(C6H5SO2)2。NF氟化剂作用制得。本品对酸稳定,血药浓度较高,组织分布广,半衰期长达8 h,并有一定的抗生素后效应,对肝脏无毒性,是一广谱低毒抗生素,且对化脓性链球菌、肺炎链球菌和金葡菌感染小鼠的保护作用优于红霉素。此外,1983年Pierrel实验室通过红霉素链霉菌(Streptomyces erythraeus ATCC31772)的生物合成制得8(S)-8-fluoroerythromycin A,B,C和D,它们均具有较好的生物活性。
, http://www.100md.com
4 C-3位酮基红霉素的结构修饰
新近Roussel Uclaf报道,将克拉霉素C-3位cladinose sugar酸催化水解后,游离出的羟基经氧化为酮基,再与二甲氨基吡啶和甲酰氯缩合环化制得3-酮基-6-氧甲基红霉素A的11,12-环状氨甲酸酯。继而对其氨基进行酰化或烷基化制得系列N-烷基化衍生物。该类衍生物既提高了对红霉素敏感菌的抗菌作用又显著增强了对红霉素耐药菌的抗菌活性。该类衍生物的成功研究否认了长期认为cladinose sugar是红霉素类抗生素活性所必需的论断。其代表衍生物为RU004和RU708。目前该实验室正从事这类衍生物的深入研究[1,2]。
此外日本大正制药公司报道的TE-802,TE-810,HMR-3004和HMR-3647等,亦属该类化合物,其中TE-810对耐药葡萄球菌、链球菌和流感杆菌均有较好抗菌活性,且能主动向白细胞与吞噬细胞移行,猴的药代动力学试验表明,其体内药代动力学优于克拉霉素。
, http://www.100md.com
综观近年来红霉素类大环内酯抗生素的结构修饰及其构效关系的研究,业已取得突破性进展,并为临床不断提供疗效卓著的红霉素新药品种。但更新的红霉素衍生物如酮基大环内酯类衍生物的深入研制和临床试验仍方兴未艾,正向纵深蓬勃发展。
参考文献
1 Georyge G,Yat SO,Daniel TW,et al.3-Keto-11,12-Carbazate derivatives of 6-O-methylerythromycin a synthesis and in vivo activity.J Antibiot,1996,49(5)∶465.
2 张致平.大环内酯类抗生素研究的进展.海口市.1996.成都:中国抗生素杂志社,1996.
(收稿:1998-11-04 修回:1999-07-05), http://www.100md.com
单位:(中国医学科学院 中国协和医科大学 医药生物技术研究所,北京 100050)
关键词:红霉素衍生物;构效关系
新红霉素衍生物的结构修饰与抗菌活性研究进展 摘要 综述了近年来红霉素衍生物的结构修饰与抗菌活性关系研究的最新进展,结果显示,该红霉素类衍生物新药品种具有组织和血药浓度高,半衰期长和抗耐药菌等药代动力学新特性。
PROGRESS IN STRUCTURE-ACTIVITY RELATIONSHIP STUDY OF
ERYTHROMYCIN DERIVATIVES
Xu Xiandong
, 百拇医药 (Institute of Medicinal Biotechnology,Peking Union Medical College,Chinese Academy of Medical Sciences,Beijing 100050)
ABSTRACT Recent progress in structure modification of erythromycin derivatives,their antibacterial activities,and pharmacokinetics were reviewed in this paper.
KEY WORDS Erythromycin derivatives;Structure-activity relationship
近20年来,根据红霉素酸缩酮化失效原理,世界药学工作者广泛深入地开展了对红霉素的结构修饰与抗菌活性研究,获得了一系列具有药代动力学新特性的红霉素类大环内酯新药品种,如罗红霉素(roxithromycin)、阿齐霉素(azithromycin)、克拉霉素(clarithromycin)、地红霉素(dirithromycin)、氟红霉素(flurithromycin)等红霉素衍生物(俗称第二代红霉素)。解决了红霉素因酸缩酮化形成8,9-脱水红霉素-6,9-半缩酮而失效,从而提高了血药浓度,延长了半衰期,改进了组织移行性,减少了用药量和副作用,增强了疗效。但这些新红霉素衍生物不仅抗菌谱、临床适应证与已知大环内酯抗生素相同,而且仍具有诱导耐药性。因此改善其耐药性,扩展其抗菌谱,增强其抗菌活性依然是该类抗生素研究的重要方向。新近Rorssel Uclaf和Abbott实验室等报道,红霉素内酯环C-3位脱cladinose sugar后转变为C-3位酮基红霉素,其衍生物的作用特点是,克服了十四元大环内酯所共有的耐药性,对大环内酯耐药菌具有较好的抗菌活性,如对耐大环内酯的肺炎球菌、金葡菌有优异的抗菌活性,对抗肠球菌活性亦增强以及对嗜血杆菌属亦有活性。
, 百拇医药
现就红霉素A分子结构中可供修饰的基团进行的结构修饰特点和抗菌活性关系作一简要综述。
1 内酯环6,11,12,和糖上2′,4″位羟基的修饰
基于内酯环上羟基的醚化,可阻止其酸性条件下分子内缩酮化形成螺缩酮,从而使其衍生物具有对酸稳定,可口服吸收快,血药浓度高而持久的特点。1981年日本大正制药公司从红霉素A出发,经肟化、醚化和硅烷化、甲基化和还原制得克拉霉素。本品对需氧菌、支原体、脲原体和衣原体等有效,体内活性比红霉素强2~4倍。又如C-6,C-11位羟基同时烷基化制得6,11-二氧烷基红霉素,其代表衍生物6,11-二甲氧基红霉素口服比红霉素A抗菌作用强4~9倍,且血药浓度高而持久。
关于内酯环上11,12位羟基同时修饰,Abbott实验室和Slawinski等曾制得一系列红霉素和克拉霉素的11,12位环状氨基甲酸酯,其代表衍生物A-66321的体外活性表明,增强了对结构耐药菌如化脓性链球菌的抗菌活性。又如Brain等在红霉素11,12位环状氨基甲酸酯的9位酮基进行肟化,同时辅以其他位置的取代制得一系列衍生物。其体内、外活性表明,对多种G+和G-菌有不同程度的抗菌活性,且未见明显毒副作用。
, http://www.100md.com
关于糖上2′,4″位羟基进行酰化制得的多种酯类衍生物如2′位乙酰酯、丙酰酯、碳酸乙酯、乙基丁二酰酯和4″-氧-乙酰胺基-6-氧甲基红霉素均已被确认为有效新型大环内酯类抗生素。
2 C-9位酮基的修饰
红霉素9位酮基最简便的修饰是变为肟基,从而阻止其分子内酸缩酮化。1972年USpatent 368326报道,利用RONH2。HX与红霉素A或B作用制得其相应肟化衍生物,并显示一定的抗菌活性。之后Jean-claude等继而对红霉素肟羟基进行醚化制得60多种醚化衍生物,经构效关系研究证明,该醚肟衍生物无论在体内、体外均具有强于红霉素A的抗菌活性,其中14种衍生物的药代动力学性质有明显改善。此外,经对肟基双键的顺反异构体的抗菌活性对照试验,发现所有顺式异构体抗菌活性较低。之后Roussel Uclaf公司通过优选法得到RU28965,即罗红霉素,并从红霉素A出发经肟化再与甲氧基乙氧基甲基氯缩合制得其产品,其组织中分布广,尤其在肺组织中浓度较高。
, 百拇医药
C-9位酮基亦可转变为胺基而制得红霉素胺(erythromycylamine),它对G+菌作用较弱,但对C-菌作用有所增强。由于其口服吸收差,后由德国Karl Thomae公司经由红霉素A胺化、重氮化、还原制得红霉素胺,再与甲氧基乙氧基乙醛缩合制得地红霉素。由于其分子结构中的C-9位胺基和11位羟基与侧链醛缩合形成
此外,Plive和Pfizer公司实验室曾将红霉素9位酮基肟化后进行Beckmann重排和N上甲基化反应,得具有十五元含氮大环内酯类抗生素阿奇霉素,本品对酸稳定,组织浓度大于血药浓度,半衰期长,其抗菌特点是对需氧和厌氧G-菌及细胞内病原菌、支原体和衣原体有较强的抗菌作用。另于1988年EP 0307128专利报道,阿齐霉素和4″位差向异构体和4″-去氧-4″-氨基类似物的抗原虫活性。Gabrijela等还报道了阿奇霉素-氧-甲基衍生物的制备,测定了它们的抗菌活性,推断了它们的构效关系。
, 百拇医药
3 C-8位质子的修饰
由于C-8位质子可用负电性强的离子或基团取代,80年代意大利Pierrel公司开发了8(S)-8-氟红霉素(flurithromycin),是将红霉素A在乙酸作用下转变为8,9-烯醇式半缩醛,再与FClO3或CF3OF和(C6H5SO2)2。NF氟化剂作用制得。本品对酸稳定,血药浓度较高,组织分布广,半衰期长达8 h,并有一定的抗生素后效应,对肝脏无毒性,是一广谱低毒抗生素,且对化脓性链球菌、肺炎链球菌和金葡菌感染小鼠的保护作用优于红霉素。此外,1983年Pierrel实验室通过红霉素链霉菌(Streptomyces erythraeus ATCC31772)的生物合成制得8(S)-8-fluoroerythromycin A,B,C和D,它们均具有较好的生物活性。
, http://www.100md.com
4 C-3位酮基红霉素的结构修饰
新近Roussel Uclaf报道,将克拉霉素C-3位cladinose sugar酸催化水解后,游离出的羟基经氧化为酮基,再与二甲氨基吡啶和甲酰氯缩合环化制得3-酮基-6-氧甲基红霉素A的11,12-环状氨甲酸酯。继而对其氨基进行酰化或烷基化制得系列N-烷基化衍生物。该类衍生物既提高了对红霉素敏感菌的抗菌作用又显著增强了对红霉素耐药菌的抗菌活性。该类衍生物的成功研究否认了长期认为cladinose sugar是红霉素类抗生素活性所必需的论断。其代表衍生物为RU004和RU708。目前该实验室正从事这类衍生物的深入研究[1,2]。
此外日本大正制药公司报道的TE-802,TE-810,HMR-3004和HMR-3647等,亦属该类化合物,其中TE-810对耐药葡萄球菌、链球菌和流感杆菌均有较好抗菌活性,且能主动向白细胞与吞噬细胞移行,猴的药代动力学试验表明,其体内药代动力学优于克拉霉素。
, http://www.100md.com
综观近年来红霉素类大环内酯抗生素的结构修饰及其构效关系的研究,业已取得突破性进展,并为临床不断提供疗效卓著的红霉素新药品种。但更新的红霉素衍生物如酮基大环内酯类衍生物的深入研制和临床试验仍方兴未艾,正向纵深蓬勃发展。
参考文献
1 Georyge G,Yat SO,Daniel TW,et al.3-Keto-11,12-Carbazate derivatives of 6-O-methylerythromycin a synthesis and in vivo activity.J Antibiot,1996,49(5)∶465.
2 张致平.大环内酯类抗生素研究的进展.海口市.1996.成都:中国抗生素杂志社,1996.
(收稿:1998-11-04 修回:1999-07-05), http://www.100md.com