ČĘÓășôłŠčÂČĄ¶Ÿ(GCRV)Čż·Ö»ùÒòÆŹ¶ÎcDNAÎÄżâ”Äččœš
ŚśŐߣș·œÇÚ ÌïŸČ
”„λŁș·œÇÚ ÌïŸČ(ÖĐčúżÆѧÔșÎäșșČĄ¶ŸŃĐŸżËùŁŹÎäșș 430071)
čŰŒüŽÊŁșČĘÓășôłŠčÂČĄ¶ŸŁ»Ë«ÁŽRNAŁ»cDNAÎÄżâ
ÖĐčúČĄ¶ŸŃ§000113 ŐȘ ÒȘŁșÔÚËæ»úÁùŸÛÌćÒęÎïĆš¶ÈČ»±äÌőŒțÏÂŁŹ·Ö±đČÉÓĂ0.5Ąą2Ąą5ŠÌg ČĘÓășôłŠčÂČĄ¶Ÿ(GCRV)”„Ò»ÆŹ¶ÎRNAœűĐĐ·ŽŚȘÂŒcDNAșÏłÉŒ°ÎÄżâččœšĄŁœáčû±íĂśŁŹÔÚGCRV-R NAÄŁ°ćĆš¶ÈŽóÓÚ2ŠÌgʱŁŹ”Ú¶țÁŽcDNAșÏłÉČúÎïżÉÖ±œÓÍščęEBÈŸÉ«”ÄÇíÖŹÌÇÄęœș”çÓŸœűĐжšÁżŒ°Œű¶šĄŁŃĄÔńcDNAÓëÔŰÌćÁŹœÓ±ÈÂÊÎȘ5ĄĂ1ŁŹÔÚžßЧ”çŚȘ»ŻÌőŒțÏÂŁŹżÉ»ń”Ă106ŚȘ»ŻŚÓ/ŠÌg cDNA”ÄŚȘ»ŻĐ§ÂÊĄŁŃôĐÔżËÂĄŚÓŸĂžÇĐŒ°PCRŒű¶šŁŹÔŒ45%ÒÔÉÏ”ÄČćÈëÆŹ¶ÎŽóÓÚ500bpĄŁ
·ÖÀàșĆŁșQ78 ÎÄÏŚ±êʶÂëŁșA
ÎÄŐ±àșĆŁș1003-5125(2000)01-0078-05
, °ÙĎҜҩ
Construction of cDNA Libraries of GCRV Partial Genome Segments
FANG Qin, TIAN Jing
(Wuhan Institute of Virology, Academia Sinica, Wuhan 430071, China)
AbstractŁșUnder the condition of constant concentration of random hex amers,the methods of cDNA synthesis and library construction were describ ed by using 0.5 ŠÌg, 2 ŠÌg and 5 ŠÌg individual RNA segment of GCRV as templates respectively. The result showed that when the template concentration of GCRV-RN A was equal to or more than 2 ŠÌg, the products of synthesized cDNA could be analysed directly through agarose gel electrophoresis stained by EB. When the ligation r atio of cDNA and vector was 5ĄĂ1, cloning efficiency could be as high as 106 transformants gene-rated per ŠÌg cDNA basing on highly efficient electropora tion transformation. Up to 45% inserts of the segments were larger than 500 bp by restriction analysis and PCR amplification.
, °ÙĎҜҩ
Key wordsŁșGrass carp reovirus (GCRV); Double-stranded RNA, cDNA libr aryĄű
ČĘÓăłöŃȘČĄČĄ¶ŸÎȘșôłŠčÂČĄ¶ŸżÆÒ»ĐÂłÉÔ±ŁŹÆä»ùÒòŚéÓÉ11ÌőË«ÁŽ·Ö¶ÎRNAŚéłÉ[1] ĄŁžùŸĘžĂČĄ¶ŸĐÎÌŹŃ§Œ°»ùÒòŚé”ÈÌŰĐÔŃĐŸżŁŹ1995ÄêčúŒÊČĄ¶Ÿ·ÖÀàĂüĂûÎŻÔ±»áÒŃŐęÊœœ«žĂČĄ¶Ÿ ĂüĂûÎȘČĘÓășôłŠčÂČĄ¶Ÿ(GCRV)ŁŹČąÄÉÈëșôłŠčÂČĄ¶ŸżÆĐÂœšËźÉúșôłŠčÂČĄ¶ŸÊôŐęÔÚŃĐŸż”ÄĐÂÖÖ [2]ŁŹÒòŽËÎÒĂÇœ«čęÈ„Ò»Ö±ŃŰÓĂ”ÄČĘÓăłöŃȘČĄČĄ¶Ÿ(GCHV)ŐęÊœžüĂûÎȘČĘÓășôłŠčÂČĄ ¶Ÿ(GCRV)ŁŹČąžùŸĘžĂČĄ¶Ÿ»ùÒòŚéRNAÔÚ”çÓŸÍŒÆŚÉϔĎóĐĄ·ÖČŒĂüĂûGCRV 11Ìő·Ö¶ÎË«ÁŽRNAÆŹ ¶Î·Ö±đÎȘL1-3ŁŹM4-6ŁŹS7-11ĄŁÄżÇ°ŁŹčúŒÊÉÏÒѱš”ÀËźÉúșôłŠčÂČĄ¶ŸÓĐ20¶àÖÖŁŹ”«¶ÔžĂÊôČĄ ¶Ÿ”ÄŃĐŸżÄÚÈĘÖśÒȘŸÖÏȚÓÚÉúÎïѧ·¶łëŁŹÎŽŒûÓĐčŰËźÉúșôłŠčÂČĄ¶Ÿ»ùÒòŚécDNAÎÄżâččœšŒ°ĐòÁĐ ·ÖÎö”ıš”ÀĄŁÎȘÁËœâËźÉúșôłŠčÂČĄ¶Ÿ»ùÒòŚéÒĆŽ«ĐĆÏąĄą»ùÒòœáččŒ°žŽÖÆÌŰŐśŁŹÓбŰÒȘœűĐĐGC RV»ùÒòŚécDNAÎÄżâ”ÄččœšĄŁŐâÒ»ŃĐŸżČ»œöÓĐÀûÓÚÁËœâËźÉúșôłŠčÂČĄ¶ŸÊô”Ä·ÖŚÓÉúÎïѧÌŰŐśŁŹ ÍŹÊ±œ«œÒÊŸËźÉúșôłŠčÂČĄ¶ŸÓëžĂżÆÆäËüÊô”ÄÇŚÔ”čŰÏ”ŁŹÒÔÈ·¶šÆäœű»ŻÀúłÌĄŁœüÆÚÎÒĂÇÒѱš”À ÁËGCRV»ùÒòŚécDNAșÏłÉĄążËÂĄŒ°Čż·ÖĐòÁĐ·ÖÎöœáčû[3]ŁŹ±ŸŃĐŸżČàÖŰÓÚÆä»ùÒòŚécD NAÎÄżâ”ÄččœšŁŹÏÖœ«œáčû±š”ÀÈçÏÂĄŁ
, °ÙĎҜҩ
1 ČÄÁÏșÍ·œ·š
1.1 ČÄÁÏ
1.1.1 ČĄ¶Ÿ¶ŸÖê
ČĘÓășôłŠčÂČĄ¶Ÿ¶ŸÖêÎȘ±ŸÊ”ŃéÊÒ±ŁŽæ¶ŸÖêGCRV873(ÔĂûGCHV873)[4] ĄŁ
1.1.2 ÊÔŒÁ
cDNAșÏłÉĄąPCRÀ©ÔöŒ°pZEr2.0żËÂĄÊÔŒÁșĐÎȘĂÀčúBRLĄąPerkin ElmerŒ°Invitrogenč«ËŸČúÆ· Ł»PCRŽż»ŻÊÔŒÁșĐŒ°ÏȚÖÆĐÔÄÚÇĐĂžÀà·Ö±đčșŚÔÉÏșŁ»ȘËŽč«ËŸŒ°»ȘĂÀÉúÎï耳Ìč«ËŸĄŁ
1.2 ·œ·š
1.2.1 GCRV»ùÒòÆŹ¶Î·ÖÀ륹Žż»Ż
, °ÙĎҜҩ
ŽÓžĐÈŸCIKÏž°ûĆàŃűÒșÖĐŽż»Ż”ÄGCRV°ŽÊ”ŃéÊÒłŁčæ·Ó/ÂÈ·ÂłéÌá·šÌáÈĄČĄ¶ŸșËËᣏGCRV-RNA ”„Ò»ÆŹ¶Î”Ä·ÖÀ댰Žż»Ż°ŽÎÄÏŚ[3]œűĐĐĄŁ
1.2.2 cDNAșÏłÉŒ°żËÂĄ
cDNAșÏłÉČÉÓĂËæ»úÒęÎï·šœűĐĐŁŹ·ŽŚȘÂŒĂžŃĄÓĂSuperScriptąòĄŁÉè¶šĂż·ŽÓŠÌć»ęÒęÎïÓĂÁżÎȘ1 00ng (25pmol/ŠÌL)ŁŹ·Ö±đČÉÓĂȻ͏”ÄÄŁ°ćÁżœűĐĐ·ŽŚȘÂŒcDNAșÏłÉĄŁŸßÌć·ŽÓŠÌőŒțŒ°·œ ·šÎȘŁșÈÜÓÚ90%DMSOÖĐ”ÄRNAŃùÆ·ŁŹÓÚ70Ąæ±äĐÔ15minŁŹÖñùÉÏŒÓÈë100ng”ÄËæ»úÁùŸÛÌć Òę ÎïŁŹÒÔĐÎłÉÄŁ°ć/ÒęÎï»ìșÏÎïĄŁÈ»șóÒÀŽÎŒÓÈë”ÚÒ»ÁŽcDNA bufferÒÔŒ°10mmol/L DTT, 0 .2u RNasin, 3ŠÌL SuperScriptąò (200u/ŠÌL)ŁŹŚîșóŒÓÈëŸDEPCŽŠÀí”ÄË«ŐôËźÖÁ100 ŠÌLĄŁ37Ąæ±ŁÎÂ60minŁŹÒÔœűĐĐcDNA”ÚÒ»ÁŽ”ÄșÏłÉĄŁcDNA”ÚÒ»ÁŽșÏłÉșóŁŹÍščęPCRŽż»Ż ĐĄÖùÈ„łęÒęÎïÁùŸÛÌ楣ČÉÓĂ0.5mol/L NaOH 37Ąæ·ŽÓŠ15minÈ„łęRNAÄŁ°ćŁŹÓĂHClÖĐșÍ șóŁŹÒÒŽŒłÁ”í·ŽÓŠČúÎïĄŁÈ»șóœ«łÁ”íÈÜÓÚ40mmol/L Tris-HCl(pH8.0)ŁŹ200mmol/L NaC l, 2mmol/L EDTA,50%ŒŚőŁ°·ÈÜÒșÖĐŁŹÓÚ94ĄæŁŹ5min±äĐÔ·ŽŚȘÂŒ”ÄÁœÌőcDNAÁŽŁŹ50 ĄæčęÒčÊčË«ÁŽcDNAÍêÈ«žŽĐÔĄŁžŽĐÔ”ÄcDNAÓĂDNA Polymerase ąń œűĐĐÁœ¶ËČčÆ룏ÒÒŽŒłÁ”íșó ŁŹÈĄ ŚÜÁż”Ä1/5”çÓŸŒű¶šŒ°łőČœ¶šÁżĄŁ¶šÁżșó”ÄcDNA°ŽČ»ÍŹ±ÈÀęÓëEcoRVÏû»Ż”ÄpZErO 2.0ÔŰÌćœű ĐĐƜͷÁŹœÓŁŹ”çŚȘ»ŻTOP 10žĐÊÜÌŹÏž°ûĄŁŚȘ»Żșó”ÄÏž°ûÍżČŒÓÚșŹKan(50ŠÌg/mL)”ÄLBÆœ°ć ÉÏŁŹčęÒčĆàŃűŁŹŽÎÈŐŒÆËăżË¥ЧÂÊĄŁÖŰŚéÖÊÁŁ”ÄĐĄÁżĆàŃűŒ°ÌáÈĄ°ŽÎÄÏŚ[5]Ìá詔ķœ·šœű ĐĐĄŁ
, http://www.100md.com
1.2.3 cDNAÎÄżâ”ÄɞѥŒ°Œű¶š
ŃĄÔńλÓÚEcoRVČćÈëÁœ¶Ë”Ä”„Ò»ĂžÇĐλ”ăBamHIșÍXhoIÏȚÖÆĐÔÄÚÇĐĂž¶ÔÖŰŚéÖÊÁŁœűĐĐË«ĂžÇĐŁŹ ŸĂžÇĐ·ÖÎöșóŁŹÌôŃĄŽóÓÚ500bp”ÄČćÈëÆŹ¶ÎœűĐĐPCRÀ©ÔöĄŁÒęÎïλ”ăÔÚŸàEcoRVČćÈëÁœ¶ËÉÏ Ïžś100bpŚóÓÒ”ÄλÖĂĄŁŐęÏòÒęÎïÎȘ5ĄäCAGGAAACAGCTATGACC 3ĄäŁ»·ŽÏòÒęÎïÎȘŁș5Ąä CGCC AGGGTTTTCCCAGTCACGAC 3ĄäĄŁÀ©ÔöÌőŒțÎȘŁș”ÚÒ»ÂÖ94Ąæ±äĐÔ3·ÖÖÓŁŹ”Ú¶țÂÖÒÔșó94Ąæ±ä ĐÔ30secŁŹ 50ĄæÍË»đ1minŁŹ70ĄæŃÓÉì2minŁŹŃ»·35ŽÎĄŁÔÚ1%ÇíÖŹÌÇÄęœș”çÓŸÌőŒț ϶ÔPCRČúÎïœűĐĐŒű¶šŒ°·ÖÎöĄŁ
2 œáčûÓëÌÖÂÛ
2.1 cDNAșÏłÉÌőŒțŒ°ČúÂÊ·ÖÎö
ÔÚč̶šĂż·ŽÓŠÌć»ęËæ»úÒęÎïÁżÎȘ100ngÌőŒțÏÂŁŹ·Ö±đČÉÓĂ500ngĄą2ŠÌgĄą5ŠÌg”Ä”„Ò» ÆŹ ¶ÎRNAœűĐĐÁËһϔÁĐGCRV-cDNAșÏłÉĄŁ·ŽŚȘÂŒ”Ú¶țÁŽcDNAșÏłÉœáčûŒûÍŒ1ĄŁŽÓÍŒ1żÉÒÔżŽłöŁŹ ÔÚRNAÄŁ°ćÁżŽóÓÚ»ò”ÈÓÚ2ŠÌgʱŁŹ”Ú¶țÁŽcDNAČúÎïŸłŁčæEBÈŸÉ«ÇíÖŹÌÇÄęœș”çÓŸșóŁŹżÉÖ± œÓÔÚŚÏÍâ”ÆÏÂœűĐĐŒű¶šŒ°¶šÁż·ÖÎöŁŹ”«”±ÄŁ°ćÓĂÁżÎȘ500ngʱcDNAČúÎïČ»ÄÜÍšč곣čæEBÈŸ É«”ÄÇíÖŹÌÇÄęœș”çÓŸœűĐĐ·ÖÎöĄŁžùŸĘ”çÓŸÍŒÆŚÉÏ”ÄGCRV-cDNAÓ«čâÇż¶ÈÓë±êŚŒMarker”Ä±ÈœÏ ·ÖÎöŁŹÍÆËă”Ú¶țÁŽcDNAșÏłÉČúÂÊÔŒÎȘŚÜÄŁ°ćÁż”Ä25%ĄŁŽËÍ⣏ŽÓÍŒÖĐ”ÄcDNAÌőŽűżÉŒûŁŹ ĐĄ·Ö ŚÓ”Ú9ÆŹ¶Î(S9)”ÄcDNA·ÖŚÓŽóĐĄÎȘ300bpĄ«1kbŁŹŽó·ÖŚÓ1Ąą3ÆŹ¶Î(L1ĄąL3)”ÄcDNAÎȘ400 bpĄ«4kbĄŁÓÉŽËżÉŒûËù”ĂcDNAČúÎï·ÖŚÓÁż·¶Î§ÓëÆäÄŁ°ćRNAŽóĐĄ»ù±ŸÎÇșÏ[1]ĄŁ
, °ÙĎҜҩ
ÍŒ1 GCRV S9ĄąL1ĄąL3ÆŹ¶Î ”ÄcDNAČúÎï”çÓŸ·ÖÎö
1. 1 kb DNA±êŚŒ·ÖŚÓÁż
2. GCRV S9”ÄcDNAČúÎï(2 ŠÌgÆđÊŒÄŁ°ć)
3. GCRV S9”ÄcDNAČúÎï(500 ngÆđÊŒÄŁ°ć)
4. GCRV L1”ÄcDNAČúÎï(5 ŠÌgÆđÊŒÄŁ°ć)
5. GCRV L3”ÄcDNAČúÎï(5 ŠÌgÆđÊŒÄŁ°ć)
6. GCHV L3”ÄcDNAČúÎï(500 ngÆđÊŒÄŁ°ć)
Fig.1 Synthesized cDNA of GCRV S9ĄąL1ĄąL3
, °ÙĎҜҩ
1. 1 kb DNA ladder
2. cDNA of GCRV S9(2 ŠÌg template)
3. cDNA of GCRV S9(500 ng template)
4. cDNA of GCRV L1(5 ŠÌg template)
5. cDNA of GCRV L3(5 ŠÌg template)
6. cDNA of GCRV L3(500 ng template)
±í1œáčûÏÔÊŸŁŹÔÚÔŰÌćÖÊÁŁDNAÓëcDNAÁŹœÓ±ÈÎȘ1ĄĂ5ʱŁŹżÉ”Ă”œžßŽï106ŚȘ»ŻŚÓ/ŠÌg cDNA”Ä ŚȘ»ŻĐ§ÂÊĄŁŐâÒ»œáčûÓë±êŚŒŠ”ĄÁ174ŚȘ»ŻĐ§ÂÊÏàČîÎȚŒž[6]ŁŹ¶űÔÚÆäËüŒžÖÖ±ÈÂÊÌő ŒțÏÂŁŹŚȘ»ŻĐ§ÂÊŸùÎŽŽï”œÔ€ÆÚЧčûĄŁžùŸĘÉÏÊöËùŃĄÔń”ÄŚîŒŃÌőŒțŁŹÎÒĂÇœűĐĐÁËGCRV-cDNA”Ä żËÂĄŁŹČą»ń”ĂÁËÀíÏë”ÄGCRV-cDNAÎĿ⣏œáčûŒû±í2ĄŁ
, °ÙĎҜҩ
±í1 cDNAÓëÔŰÌćÆœÁŹ±ÈÂÊ¶ÔŚȘ»ŻĐ§ÂÊ”ÄÓ°Ïì
Table 1 Effect of the ratio of blunt-end ligation of
cDNA and vector on transformation efficiency Exp.
No.
Vector
(ng)
cDNA
(ng)
Plasmid/cDNA
approx. ratio
, http://www.100md.com
Efficiency
(transformants/ŠÌg cDNA)
1
50
-
-
0
2
50
50
1ĄĂ1
5.0ĄÁ103
3
, http://www.100md.com
50
100
1ĄĂ2
8.9ĄÁ104
4
50
250
1ĄĂ5
4.8ĄÁ106
5
50
500
1ĄĂ10
, °ÙĎҜҩ
4.5ĄÁ104
±í2 cDNAżËÂĄŒ°ÎÄżâÌŰĐÔ
Table 2 cDNA cloning and library properties Library construction
Library 1 (L1)
Librar y 6 (M6)
Library 10 (S10)
RNA template size(bp)
4800
1900
890
, °ÙĎҜҩ
RNA template input(ŠÌg)
5
2
2
Library size (Nb clones)
4.9ĄÁ106
5.2ĄÁ106
4.1ĄÁ106
Insert analysis
Number of analysed clones
, °ÙĎҜҩ 100
Ration
100
Ration
100
Ration
Number of inserts>500 bp
62
62%
56
56%
45
45%
, °ÙĎҜҩ
Background
3
3%
2
2%
2
2%
2.3 ŃôĐÔżËÂĄ”ÄŒű¶š
ɞѥ”ÄÖŰŚéÖÊÁŁÊŚÏÈÓĂĂžÇĐ·šœűĐĐłőČœŒű¶šŁŹÈ»șóžùŸĘÖÊÁŁÔŰÌćÉÏ”ÄÒęÎïλ”ăœűĐĐPCRÀ©Ôö ŁŹËù”ĂÀ©ÔöČúÎïŸÄęœș”çÓŸ·ÖÎöŁŹÆäÀ©ÔöÆŹ¶Î”Ä·ÖŚÓÁżŽóĐĄÓëĂžÇĐœáčûÏàÎÇșÏ(ÍŒ2AŁŹB)ĄŁ ÍŒ2AÎȘL1ÆŹ¶ÎÖŰŚéżËÂĄ”ÄË«ĂžÇĐÍŒÆŚŁ»ÍŒ2BÎȘÓë2A(1-9)Ïà¶ÔÓŠ”ÄPCRÀ©ÔöČúÎïĄŁÆä2BžśÀ© ÔöÆŹ¶Î”Ä·ÖŚÓÁżŸùŽóÓÚĂžÇĐÆŹ¶Î200bpŚóÓÒŁŹŐęșĂÓëÔ€Æڔķ֌ÓÁżŽóĐĄÏàÒ»ÖÂĄŁ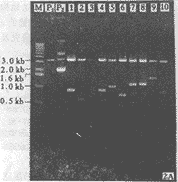
, http://www.100md.com
ÍŒ2A ÖŰŚéÖÊÁŁ”ÄĂžÇĐ·ÖÎö
M. 1kb DNA±êŚŒ·ÖŚÓÁż
P1.ÏßĐÔ»Ż”ÄpZErO2.0ÖÊÁŁ
P2.łŹÂĘĐęșÍÏßĐÔ»Ż”ÄpZErO2.0ÖÊÁŁ
1-10.BamHIșÍXholIÏû»Ż”ÄL1ÆŹ¶Î”ÄÖŰŚéÖÊÁŁ
Fig.2A Restriction analysis of GCRV L1
M. 1 kb DNA ladder
P1. Linearized pZErO2.0
P2. Supercoiled and linearized pZErO2.0
, http://www.100md.com
1-10. Digested recombinant plasmids of L1
by BamHI and XholI
ÍŒ2B GCRV L1ÆŹ¶ÎÖŰŚéÖÊÁŁ”ÄPCRÀ©ÔöČúÎï·ÖÎö
M. 1 kb DNA±êŚŒ·ÖŚÓÁż
N.ÒőĐÔ¶ÔŐŐ
1-9. L1ÆŹ¶Î”ÄPCRÀ©ÔöČúÎï
Fig.2B PCR amplification of recombinant
plasmids of GCRV L1
, °ÙĎҜҩ
M. 1 kb DNA ladder
N. Negative control
1-9. PCR amplification products of GCRV L1
3 ÌÖÂÛ
ŚÔ1983ÄêÊŚŽÎ·ÖÀëGCRVŁŹÆùœńÒŃÓĐÊź¶àÄê”ÄÀúÊ·ĄŁÓÉÓÚGCRVșŹ11ÌőË«ÁŽ·Ö¶ÎRNA»ùÒòŚéÒÔŒ° Æä3Ąä¶ËČ»șŹpoly(A)ÎČ”ÄÌŰĐÔŁŹÊč”ĂÆä»ùÒòŚéÎÄżâ”ÄččœšŸßÓĐÒ»¶š”ÄÄŃ¶ÈŒ°žŽÔÓĐÔĄŁÔçÆÚșô łŠčÂČĄ¶ŸcDNAÎÄżâ”ÄččœšÖśÒȘœèÖúŽ«Íł”ÄcDNAșÏłÉŒ°żËÂĄ·œ·šŁŹÆäÖśÒȘŒŒÊő»ù±ŸÂ·ÏßÎȘŁșÍš čępoly(A)ŒÓÎČ·ŽÓŠ»ń”ĂŽűpoly(A)ÎČ”ÄË«ÁŽRNAŁŹŒÌ¶űČÉÓĂolig (dT)Òę”ŒcDNA”ÚÒ»ÁŽșÏłÉĄŁ žùŸĘCashdollar”ÈÈË[7]”ıš”ÀŁŹÔÚœűĐĐșôłŠčÂČĄ¶ŸąóĐÍ(strain Dearing) cDNA żËÂĄÊ±ŁŹÔűÓĂ150ŠÌgŸÍŹÎ»ËŰ(3HÄòàŚà€)±êŒÇ”ÄÈ«»ùÒòŚéRNAœűĐĐpoly(A)ŒÓÎČ·ŽÓŠ ŁŹÔÚcDNAșÏłÉʱŁŹÈÔÓĂ32P-dATPÔَαêŒÇcDNA”ÚÒ»ÁŽșÏłÉĄŁÈçŽËŽó”ÄÄŁ°ćÓĂÁż Œ°ł€Ê±Œä”Ä͏λËŰČÙŚśŁŹÔÚÒ»°ăÊ”ŃéÌőŒțÏÂÊÇÄŃÒÔœűĐĐșÍżŰÖÆ”ÄĄŁœüÄêÀŽ”ÄŚÊÁÏŒ°ÎÄÏŚÏÔÊŸ ŁŹčúÍâ¶ÔșôłŠčÂČĄ¶ŸżÆÔÓĐÁùžöÊô”Ä·ÖŚÓÉúÎïѧÌŰĐÔÒŃŃĐŸż”ĂÏà”±ÉîÈ룏ȻœöÓĐÎÄżâččœšŒ° »ùÒòŚéÆŹ¶Î”ÄÈ«ĐòÁбš”À[8ŁŹ9]ŁŹ¶űÇÒ¶ÔijЩÖŰÒȘ»ùÒòÆŹ¶Î”ÄœáččÓëčŠÄÜŒ°ŚȘÂŒ ”śżŰ·œĂæ”ÄŃĐŸżÒČÒŃÆđČœ[10ŁŹ11]ĄŁËäÈ»ÄżÇ°ŚîÎȘÆŐ±é”ÄcDNAÎÄżâččœš¶àČÉÓĂ RT-PCRșÏłÉcDNAŁŹ”«žĂ·œ·šŸùĐèœèÖúŒÆËă»úÊęŸĘżâœűĐĐÏàčŰÖÖÊôĐòÁĐ”ÄÍŹÔŽĐÔŒ°±ŁÊŰÇűÓò ”Ä·ÖÎöŁŹÒÔ»ń”ĂRT-PCR”ÄÒęÎïĐòÁĐŁŹŽÓ¶űččœšÍŹÀàČĄ¶ŸĐ¶ŸÖê”ÄcDNAÎĿ⥣ÓÉÓÚGCRVÎȘșô łŠčÂČĄ¶ŸżÆĐÂÊôĐÂÖÖŁŹÔÚÎȚ·šœűĐĐÍŹÔŽĐòÁĐ±ÈœÏ”ÄÇéżöÏÂŁŹČÉÓĂËæ»úÒęÎï·šÒę”ŒcDNAșÏłÉČ» ʧÎȘÒ»ÖÖŒò”„ĄążÉĐĐ”ÄČßÂÔĄŁÎÒĂÇÔÚÔÓĐËæ»úÒęÎï·š”Ä»ùŽĄÉÏŁŹÍščęœűĐĐһϔÁĐÌőŒț±ÈœÏŁŹ ČÉÓĂÎążËŒ¶ÄŁ°ćŁŹłÉ芔ŰččœšÁËGCRV-RNA L1ĄąL2ĄąL3ĄąM6ĄąS7ĄąS9ĄąS10ĄąS11”ÄcDNAÎÄżâ ĄŁÄżÇ°ÎÒĂÇŐęÀûÓĂËùččœš”ÄcDNAÎÄżâœűĐĐGCRVžś»ùÒòÆŹ¶Î”ÄĐòÁĐ·ÖÎöŁŹŐ✫ÎȘžĂČĄ¶ŸÒÔșó”Ä ÍŹÔŽĐÔ±ÈœÏĄąœű»ŻŃĐŸżŒ°žś»ùÒòÆŹ¶ÎËù±àÂë”Ä”°°ŚÖÊœáččÓëčŠÄÜŃĐŸż”춚ÁŒșÔĻùŽĄĄŁĄö
, °ÙĎҜҩ
»ùœđÏîÄżŁșčúŒÒŚÔÈ»żÆѧ»ùœđŚÊÖúżÎÌâ ĆúŚŒșĆŁș39570035
ŚśŐߌòœéŁș·œÇÚ(1961-)ŁŹĆźŁŹșț±±ÊĄÈËŁŹž±ŃĐŸżÔ±ĄŁÖśÒȘŽÓÊÂËźÉú¶ŻÎïČĄ¶Ÿ”Ä·ÖŚÓ ÉúÎïѧŒ°Æä»ùÒò耳ÌŃĐŸżŁŹłĐ”ŁčęčúŒÒŚÔÈ»żÆѧ»ùÒòÏîÄżŁŹ·ą±íÂÛÎÄ20ÓàÆȘĄŁ
ČÎżŒÎÄÏŚŁș
[1]żÂÀö»ȘŁŹ·œÇÚŁŹČÌÒËÈš.Ò»ÖêĐ”ÄČĘÓăłöŃȘČĄČĄ¶Ÿ·ÖÀëÎï”ÄÌŰĐÔ[J].ËźÉúÉúÎïѧ±šŁŹ1990ŁŹ14(2)Łș153Ą«159
[2]Murphy F A, Fauquet C M, Bishop D H L et al. Virus Taxnomy [J]. Arc hives of Virology, 1995,(suppl):208Ą«239
[3]ÌïŸČŁŹŚȚčđÆœŁŹ·œÇÚ.ČĘÓăłöŃȘČĄČĄ¶Ÿ»ùÒòŚé”ÄcDNAșÏłÉĄążËÂĄŒ°ĐòÁĐ·ÖÎö[J].ÖĐčúČĄ¶ŸŃ§ŁŹ1999ŁŹ14Łș87Ą«92
, http://www.100md.com
[4]·œÇÚŁŹżÂÀö»ȘŁŹČÌÒËÈš.ČĘÓăłöŃȘČĄČĄ¶Ÿ”ÄÉúł€ÌŰĐÔŒ°žß”ÎŒÛĆàŃű[J].ČĄ¶ŸŃ§ÔÓÖŸ , 1989,4(3):315Ą«319
[5]Sambrook J, Fritsch E F, Maniatis T. Molecular Cloning, A Laboratory Manu al [M], 2nd ed. USA, NY: Cold Spring Harbor, 1989.16Ą«23
[6]·œÇÚŁŹÌïŸČ.”çŚȘ»Ż·šÌážßÆœÁŹÔŰÌćDNAŚȘ»ŻĐ§ÂÊ”ÄŃĐŸż[J].ÉúÎÊőŁŹ1999ŁŹ9(4)Łș5Ą«8
[7]Cashdollar L W, Esparza, Hudson G R et al. Cloning the double-strand RNA genes of reovirus: Sequence of the cloned S2 gene [J]. Biochemistry, 1982 ,79:7644Ą«7648
, °ÙĎҜҩ
[8]Wiener J R, Joklik W K. The Sequences of the reovirus serotype 1, 2, and 3 L1 genome segments and analysis of the mode of divergence of the reovirus sero types [J]. Virology, 1989,169:194Ą«203
[9]Patton J T, Salter-CID L Kalbach A et al. Nucleotide and amino acid sequence analysis of the rotavirus non-structural RNA-binding protein NS35 [ J]. Virology, 1993,192:438Ą«446
[10]Lu G Y, Zhou Z H, Baker M L et al. Structure of double-shelle d rice dwarf virus [J].J Virol, 1998,72:8541Ą«8549
[11]Schiff L A, Nibert M L, Co M S et al. Distinct binding sites for zinc and double-stranded RNA in the reovirus outer capsid protein ŠÄ3[J]. Mol Cell Biol, 1988,8:273Ą«283
ÊŐžćÈŐÆÚŁș1999-05-21
ĐȚžćÈŐÆÚŁș1999-08-16, http://www.100md.com
”„λŁș·œÇÚ ÌïŸČ(ÖĐčúżÆѧÔșÎäșșČĄ¶ŸŃĐŸżËùŁŹÎäșș 430071)
čŰŒüŽÊŁșČĘÓășôłŠčÂČĄ¶ŸŁ»Ë«ÁŽRNAŁ»cDNAÎÄżâ
ÖĐčúČĄ¶ŸŃ§000113 ŐȘ ÒȘŁșÔÚËæ»úÁùŸÛÌćÒęÎïĆš¶ÈČ»±äÌőŒțÏÂŁŹ·Ö±đČÉÓĂ0.5Ąą2Ąą5ŠÌg ČĘÓășôłŠčÂČĄ¶Ÿ(GCRV)”„Ò»ÆŹ¶ÎRNAœűĐĐ·ŽŚȘÂŒcDNAșÏłÉŒ°ÎÄżâččœšĄŁœáčû±íĂśŁŹÔÚGCRV-R NAÄŁ°ćĆš¶ÈŽóÓÚ2ŠÌgʱŁŹ”Ú¶țÁŽcDNAșÏłÉČúÎïżÉÖ±œÓÍščęEBÈŸÉ«”ÄÇíÖŹÌÇÄęœș”çÓŸœűĐжšÁżŒ°Œű¶šĄŁŃĄÔńcDNAÓëÔŰÌćÁŹœÓ±ÈÂÊÎȘ5ĄĂ1ŁŹÔÚžßЧ”çŚȘ»ŻÌőŒțÏÂŁŹżÉ»ń”Ă106ŚȘ»ŻŚÓ/ŠÌg cDNA”ÄŚȘ»ŻĐ§ÂÊĄŁŃôĐÔżËÂĄŚÓŸĂžÇĐŒ°PCRŒű¶šŁŹÔŒ45%ÒÔÉÏ”ÄČćÈëÆŹ¶ÎŽóÓÚ500bpĄŁ
·ÖÀàșĆŁșQ78 ÎÄÏŚ±êʶÂëŁșA
ÎÄŐ±àșĆŁș1003-5125(2000)01-0078-05
, °ÙĎҜҩ
Construction of cDNA Libraries of GCRV Partial Genome Segments
FANG Qin, TIAN Jing
(Wuhan Institute of Virology, Academia Sinica, Wuhan 430071, China)
AbstractŁșUnder the condition of constant concentration of random hex amers,the methods of cDNA synthesis and library construction were describ ed by using 0.5 ŠÌg, 2 ŠÌg and 5 ŠÌg individual RNA segment of GCRV as templates respectively. The result showed that when the template concentration of GCRV-RN A was equal to or more than 2 ŠÌg, the products of synthesized cDNA could be analysed directly through agarose gel electrophoresis stained by EB. When the ligation r atio of cDNA and vector was 5ĄĂ1, cloning efficiency could be as high as 106 transformants gene-rated per ŠÌg cDNA basing on highly efficient electropora tion transformation. Up to 45% inserts of the segments were larger than 500 bp by restriction analysis and PCR amplification.
, °ÙĎҜҩ
Key wordsŁșGrass carp reovirus (GCRV); Double-stranded RNA, cDNA libr aryĄű
ČĘÓăłöŃȘČĄČĄ¶ŸÎȘșôłŠčÂČĄ¶ŸżÆÒ»ĐÂłÉÔ±ŁŹÆä»ùÒòŚéÓÉ11ÌőË«ÁŽ·Ö¶ÎRNAŚéłÉ[1] ĄŁžùŸĘžĂČĄ¶ŸĐÎÌŹŃ§Œ°»ùÒòŚé”ÈÌŰĐÔŃĐŸżŁŹ1995ÄêčúŒÊČĄ¶Ÿ·ÖÀàĂüĂûÎŻÔ±»áÒŃŐęÊœœ«žĂČĄ¶Ÿ ĂüĂûÎȘČĘÓășôłŠčÂČĄ¶Ÿ(GCRV)ŁŹČąÄÉÈëșôłŠčÂČĄ¶ŸżÆĐÂœšËźÉúșôłŠčÂČĄ¶ŸÊôŐęÔÚŃĐŸż”ÄĐÂÖÖ [2]ŁŹÒòŽËÎÒĂÇœ«čęÈ„Ò»Ö±ŃŰÓĂ”ÄČĘÓăłöŃȘČĄČĄ¶Ÿ(GCHV)ŐęÊœžüĂûÎȘČĘÓășôłŠčÂČĄ ¶Ÿ(GCRV)ŁŹČąžùŸĘžĂČĄ¶Ÿ»ùÒòŚéRNAÔÚ”çÓŸÍŒÆŚÉϔĎóĐĄ·ÖČŒĂüĂûGCRV 11Ìő·Ö¶ÎË«ÁŽRNAÆŹ ¶Î·Ö±đÎȘL1-3ŁŹM4-6ŁŹS7-11ĄŁÄżÇ°ŁŹčúŒÊÉÏÒѱš”ÀËźÉúșôłŠčÂČĄ¶ŸÓĐ20¶àÖÖŁŹ”«¶ÔžĂÊôČĄ ¶Ÿ”ÄŃĐŸżÄÚÈĘÖśÒȘŸÖÏȚÓÚÉúÎïѧ·¶łëŁŹÎŽŒûÓĐčŰËźÉúșôłŠčÂČĄ¶Ÿ»ùÒòŚécDNAÎÄżâččœšŒ°ĐòÁĐ ·ÖÎö”ıš”ÀĄŁÎȘÁËœâËźÉúșôłŠčÂČĄ¶Ÿ»ùÒòŚéÒĆŽ«ĐĆÏąĄą»ùÒòœáččŒ°žŽÖÆÌŰŐśŁŹÓбŰÒȘœűĐĐGC RV»ùÒòŚécDNAÎÄżâ”ÄččœšĄŁŐâÒ»ŃĐŸżČ»œöÓĐÀûÓÚÁËœâËźÉúșôłŠčÂČĄ¶ŸÊô”Ä·ÖŚÓÉúÎïѧÌŰŐśŁŹ ÍŹÊ±œ«œÒÊŸËźÉúșôłŠčÂČĄ¶ŸÓëžĂżÆÆäËüÊô”ÄÇŚÔ”čŰÏ”ŁŹÒÔÈ·¶šÆäœű»ŻÀúłÌĄŁœüÆÚÎÒĂÇÒѱš”À ÁËGCRV»ùÒòŚécDNAșÏłÉĄążËÂĄŒ°Čż·ÖĐòÁĐ·ÖÎöœáčû[3]ŁŹ±ŸŃĐŸżČàÖŰÓÚÆä»ùÒòŚécD NAÎÄżâ”ÄččœšŁŹÏÖœ«œáčû±š”ÀÈçÏÂĄŁ
, °ÙĎҜҩ
1 ČÄÁÏșÍ·œ·š
1.1 ČÄÁÏ
1.1.1 ČĄ¶Ÿ¶ŸÖê
ČĘÓășôłŠčÂČĄ¶Ÿ¶ŸÖêÎȘ±ŸÊ”ŃéÊÒ±ŁŽæ¶ŸÖêGCRV873(ÔĂûGCHV873)[4] ĄŁ
1.1.2 ÊÔŒÁ
cDNAșÏłÉĄąPCRÀ©ÔöŒ°pZEr2.0żËÂĄÊÔŒÁșĐÎȘĂÀčúBRLĄąPerkin ElmerŒ°Invitrogenč«ËŸČúÆ· Ł»PCRŽż»ŻÊÔŒÁșĐŒ°ÏȚÖÆĐÔÄÚÇĐĂžÀà·Ö±đčșŚÔÉÏșŁ»ȘËŽč«ËŸŒ°»ȘĂÀÉúÎï耳Ìč«ËŸĄŁ
1.2 ·œ·š
1.2.1 GCRV»ùÒòÆŹ¶Î·ÖÀ륹Žż»Ż
, °ÙĎҜҩ
ŽÓžĐÈŸCIKÏž°ûĆàŃűÒșÖĐŽż»Ż”ÄGCRV°ŽÊ”ŃéÊÒłŁčæ·Ó/ÂÈ·ÂłéÌá·šÌáÈĄČĄ¶ŸșËËᣏGCRV-RNA ”„Ò»ÆŹ¶Î”Ä·ÖÀ댰Žż»Ż°ŽÎÄÏŚ[3]œűĐĐĄŁ
1.2.2 cDNAșÏłÉŒ°żËÂĄ
cDNAșÏłÉČÉÓĂËæ»úÒęÎï·šœűĐĐŁŹ·ŽŚȘÂŒĂžŃĄÓĂSuperScriptąòĄŁÉè¶šĂż·ŽÓŠÌć»ęÒęÎïÓĂÁżÎȘ1 00ng (25pmol/ŠÌL)ŁŹ·Ö±đČÉÓĂȻ͏”ÄÄŁ°ćÁżœűĐĐ·ŽŚȘÂŒcDNAșÏłÉĄŁŸßÌć·ŽÓŠÌőŒțŒ°·œ ·šÎȘŁșÈÜÓÚ90%DMSOÖĐ”ÄRNAŃùÆ·ŁŹÓÚ70Ąæ±äĐÔ15minŁŹÖñùÉÏŒÓÈë100ng”ÄËæ»úÁùŸÛÌć Òę ÎïŁŹÒÔĐÎłÉÄŁ°ć/ÒęÎï»ìșÏÎïĄŁÈ»șóÒÀŽÎŒÓÈë”ÚÒ»ÁŽcDNA bufferÒÔŒ°10mmol/L DTT, 0 .2u RNasin, 3ŠÌL SuperScriptąò (200u/ŠÌL)ŁŹŚîșóŒÓÈëŸDEPCŽŠÀí”ÄË«ŐôËźÖÁ100 ŠÌLĄŁ37Ąæ±ŁÎÂ60minŁŹÒÔœűĐĐcDNA”ÚÒ»ÁŽ”ÄșÏłÉĄŁcDNA”ÚÒ»ÁŽșÏłÉșóŁŹÍščęPCRŽż»Ż ĐĄÖùÈ„łęÒęÎïÁùŸÛÌ楣ČÉÓĂ0.5mol/L NaOH 37Ąæ·ŽÓŠ15minÈ„łęRNAÄŁ°ćŁŹÓĂHClÖĐșÍ șóŁŹÒÒŽŒłÁ”í·ŽÓŠČúÎïĄŁÈ»șóœ«łÁ”íÈÜÓÚ40mmol/L Tris-HCl(pH8.0)ŁŹ200mmol/L NaC l, 2mmol/L EDTA,50%ŒŚőŁ°·ÈÜÒșÖĐŁŹÓÚ94ĄæŁŹ5min±äĐÔ·ŽŚȘÂŒ”ÄÁœÌőcDNAÁŽŁŹ50 ĄæčęÒčÊčË«ÁŽcDNAÍêÈ«žŽĐÔĄŁžŽĐÔ”ÄcDNAÓĂDNA Polymerase ąń œűĐĐÁœ¶ËČčÆ룏ÒÒŽŒłÁ”íșó ŁŹÈĄ ŚÜÁż”Ä1/5”çÓŸŒű¶šŒ°łőČœ¶šÁżĄŁ¶šÁżșó”ÄcDNA°ŽČ»ÍŹ±ÈÀęÓëEcoRVÏû»Ż”ÄpZErO 2.0ÔŰÌćœű ĐĐƜͷÁŹœÓŁŹ”çŚȘ»ŻTOP 10žĐÊÜÌŹÏž°ûĄŁŚȘ»Żșó”ÄÏž°ûÍżČŒÓÚșŹKan(50ŠÌg/mL)”ÄLBÆœ°ć ÉÏŁŹčęÒčĆàŃűŁŹŽÎÈŐŒÆËăżË¥ЧÂÊĄŁÖŰŚéÖÊÁŁ”ÄĐĄÁżĆàŃűŒ°ÌáÈĄ°ŽÎÄÏŚ[5]Ìá詔ķœ·šœű ĐĐĄŁ
, http://www.100md.com
1.2.3 cDNAÎÄżâ”ÄɞѥŒ°Œű¶š
ŃĄÔńλÓÚEcoRVČćÈëÁœ¶Ë”Ä”„Ò»ĂžÇĐλ”ăBamHIșÍXhoIÏȚÖÆĐÔÄÚÇĐĂž¶ÔÖŰŚéÖÊÁŁœűĐĐË«ĂžÇĐŁŹ ŸĂžÇĐ·ÖÎöșóŁŹÌôŃĄŽóÓÚ500bp”ÄČćÈëÆŹ¶ÎœűĐĐPCRÀ©ÔöĄŁÒęÎïλ”ăÔÚŸàEcoRVČćÈëÁœ¶ËÉÏ Ïžś100bpŚóÓÒ”ÄλÖĂĄŁŐęÏòÒęÎïÎȘ5ĄäCAGGAAACAGCTATGACC 3ĄäŁ»·ŽÏòÒęÎïÎȘŁș5Ąä CGCC AGGGTTTTCCCAGTCACGAC 3ĄäĄŁÀ©ÔöÌőŒțÎȘŁș”ÚÒ»ÂÖ94Ąæ±äĐÔ3·ÖÖÓŁŹ”Ú¶țÂÖÒÔșó94Ąæ±ä ĐÔ30secŁŹ 50ĄæÍË»đ1minŁŹ70ĄæŃÓÉì2minŁŹŃ»·35ŽÎĄŁÔÚ1%ÇíÖŹÌÇÄęœș”çÓŸÌőŒț ϶ÔPCRČúÎïœűĐĐŒű¶šŒ°·ÖÎöĄŁ
2 œáčûÓëÌÖÂÛ
2.1 cDNAșÏłÉÌőŒțŒ°ČúÂÊ·ÖÎö
ÔÚč̶šĂż·ŽÓŠÌć»ęËæ»úÒęÎïÁżÎȘ100ngÌőŒțÏÂŁŹ·Ö±đČÉÓĂ500ngĄą2ŠÌgĄą5ŠÌg”Ä”„Ò» ÆŹ ¶ÎRNAœűĐĐÁËһϔÁĐGCRV-cDNAșÏłÉĄŁ·ŽŚȘÂŒ”Ú¶țÁŽcDNAșÏłÉœáčûŒûÍŒ1ĄŁŽÓÍŒ1żÉÒÔżŽłöŁŹ ÔÚRNAÄŁ°ćÁżŽóÓÚ»ò”ÈÓÚ2ŠÌgʱŁŹ”Ú¶țÁŽcDNAČúÎïŸłŁčæEBÈŸÉ«ÇíÖŹÌÇÄęœș”çÓŸșóŁŹżÉÖ± œÓÔÚŚÏÍâ”ÆÏÂœűĐĐŒű¶šŒ°¶šÁż·ÖÎöŁŹ”«”±ÄŁ°ćÓĂÁżÎȘ500ngʱcDNAČúÎïČ»ÄÜÍšč곣čæEBÈŸ É«”ÄÇíÖŹÌÇÄęœș”çÓŸœűĐĐ·ÖÎöĄŁžùŸĘ”çÓŸÍŒÆŚÉÏ”ÄGCRV-cDNAÓ«čâÇż¶ÈÓë±êŚŒMarker”Ä±ÈœÏ ·ÖÎöŁŹÍÆËă”Ú¶țÁŽcDNAșÏłÉČúÂÊÔŒÎȘŚÜÄŁ°ćÁż”Ä25%ĄŁŽËÍ⣏ŽÓÍŒÖĐ”ÄcDNAÌőŽűżÉŒûŁŹ ĐĄ·Ö ŚÓ”Ú9ÆŹ¶Î(S9)”ÄcDNA·ÖŚÓŽóĐĄÎȘ300bpĄ«1kbŁŹŽó·ÖŚÓ1Ąą3ÆŹ¶Î(L1ĄąL3)”ÄcDNAÎȘ400 bpĄ«4kbĄŁÓÉŽËżÉŒûËù”ĂcDNAČúÎï·ÖŚÓÁż·¶Î§ÓëÆäÄŁ°ćRNAŽóĐĄ»ù±ŸÎÇșÏ[1]ĄŁ
, °ÙĎҜҩ
ÍŒ1 GCRV S9ĄąL1ĄąL3ÆŹ¶Î ”ÄcDNAČúÎï”çÓŸ·ÖÎö
1. 1 kb DNA±êŚŒ·ÖŚÓÁż
2. GCRV S9”ÄcDNAČúÎï(2 ŠÌgÆđÊŒÄŁ°ć)
3. GCRV S9”ÄcDNAČúÎï(500 ngÆđÊŒÄŁ°ć)
4. GCRV L1”ÄcDNAČúÎï(5 ŠÌgÆđÊŒÄŁ°ć)
5. GCRV L3”ÄcDNAČúÎï(5 ŠÌgÆđÊŒÄŁ°ć)
6. GCHV L3”ÄcDNAČúÎï(500 ngÆđÊŒÄŁ°ć)
Fig.1 Synthesized cDNA of GCRV S9ĄąL1ĄąL3
, °ÙĎҜҩ
1. 1 kb DNA ladder
2. cDNA of GCRV S9(2 ŠÌg template)
3. cDNA of GCRV S9(500 ng template)
4. cDNA of GCRV L1(5 ŠÌg template)
5. cDNA of GCRV L3(5 ŠÌg template)
6. cDNA of GCRV L3(500 ng template)
±í1œáčûÏÔÊŸŁŹÔÚÔŰÌćÖÊÁŁDNAÓëcDNAÁŹœÓ±ÈÎȘ1ĄĂ5ʱŁŹżÉ”Ă”œžßŽï106ŚȘ»ŻŚÓ/ŠÌg cDNA”Ä ŚȘ»ŻĐ§ÂÊĄŁŐâÒ»œáčûÓë±êŚŒŠ”ĄÁ174ŚȘ»ŻĐ§ÂÊÏàČîÎȚŒž[6]ŁŹ¶űÔÚÆäËüŒžÖÖ±ÈÂÊÌő ŒțÏÂŁŹŚȘ»ŻĐ§ÂÊŸùÎŽŽï”œÔ€ÆÚЧčûĄŁžùŸĘÉÏÊöËùŃĄÔń”ÄŚîŒŃÌőŒțŁŹÎÒĂÇœűĐĐÁËGCRV-cDNA”Ä żËÂĄŁŹČą»ń”ĂÁËÀíÏë”ÄGCRV-cDNAÎĿ⣏œáčûŒû±í2ĄŁ
, °ÙĎҜҩ
±í1 cDNAÓëÔŰÌćÆœÁŹ±ÈÂÊ¶ÔŚȘ»ŻĐ§ÂÊ”ÄÓ°Ïì
Table 1 Effect of the ratio of blunt-end ligation of
cDNA and vector on transformation efficiency Exp.
No.
Vector
(ng)
cDNA
(ng)
Plasmid/cDNA
approx. ratio
, http://www.100md.com
Efficiency
(transformants/ŠÌg cDNA)
1
50
-
-
0
2
50
50
1ĄĂ1
5.0ĄÁ103
3
, http://www.100md.com
50
100
1ĄĂ2
8.9ĄÁ104
4
50
250
1ĄĂ5
4.8ĄÁ106
5
50
500
1ĄĂ10
, °ÙĎҜҩ
4.5ĄÁ104
±í2 cDNAżËÂĄŒ°ÎÄżâÌŰĐÔ
Table 2 cDNA cloning and library properties Library construction
Library 1 (L1)
Librar y 6 (M6)
Library 10 (S10)
RNA template size(bp)
4800
1900
890
, °ÙĎҜҩ
RNA template input(ŠÌg)
5
2
2
Library size (Nb clones)
4.9ĄÁ106
5.2ĄÁ106
4.1ĄÁ106
Insert analysis
Number of analysed clones
, °ÙĎҜҩ 100
Ration
100
Ration
100
Ration
Number of inserts>500 bp
62
62%
56
56%
45
45%
, °ÙĎҜҩ
Background
3
3%
2
2%
2
2%
2.3 ŃôĐÔżËÂĄ”ÄŒű¶š
ɞѥ”ÄÖŰŚéÖÊÁŁÊŚÏÈÓĂĂžÇĐ·šœűĐĐłőČœŒű¶šŁŹÈ»șóžùŸĘÖÊÁŁÔŰÌćÉÏ”ÄÒęÎïλ”ăœűĐĐPCRÀ©Ôö ŁŹËù”ĂÀ©ÔöČúÎïŸÄęœș”çÓŸ·ÖÎöŁŹÆäÀ©ÔöÆŹ¶Î”Ä·ÖŚÓÁżŽóĐĄÓëĂžÇĐœáčûÏàÎÇșÏ(ÍŒ2AŁŹB)ĄŁ ÍŒ2AÎȘL1ÆŹ¶ÎÖŰŚéżËÂĄ”ÄË«ĂžÇĐÍŒÆŚŁ»ÍŒ2BÎȘÓë2A(1-9)Ïà¶ÔÓŠ”ÄPCRÀ©ÔöČúÎïĄŁÆä2BžśÀ© ÔöÆŹ¶Î”Ä·ÖŚÓÁżŸùŽóÓÚĂžÇĐÆŹ¶Î200bpŚóÓÒŁŹŐęșĂÓëÔ€Æڔķ֌ÓÁżŽóĐĄÏàÒ»ÖÂĄŁ
, http://www.100md.com
ÍŒ2A ÖŰŚéÖÊÁŁ”ÄĂžÇĐ·ÖÎö
M. 1kb DNA±êŚŒ·ÖŚÓÁż
P1.ÏßĐÔ»Ż”ÄpZErO2.0ÖÊÁŁ
P2.łŹÂĘĐęșÍÏßĐÔ»Ż”ÄpZErO2.0ÖÊÁŁ
1-10.BamHIșÍXholIÏû»Ż”ÄL1ÆŹ¶Î”ÄÖŰŚéÖÊÁŁ
Fig.2A Restriction analysis of GCRV L1
M. 1 kb DNA ladder
P1. Linearized pZErO2.0
P2. Supercoiled and linearized pZErO2.0
, http://www.100md.com
1-10. Digested recombinant plasmids of L1
by BamHI and XholI
ÍŒ2B GCRV L1ÆŹ¶ÎÖŰŚéÖÊÁŁ”ÄPCRÀ©ÔöČúÎï·ÖÎö
M. 1 kb DNA±êŚŒ·ÖŚÓÁż
N.ÒőĐÔ¶ÔŐŐ
1-9. L1ÆŹ¶Î”ÄPCRÀ©ÔöČúÎï
Fig.2B PCR amplification of recombinant
plasmids of GCRV L1
, °ÙĎҜҩ
M. 1 kb DNA ladder
N. Negative control
1-9. PCR amplification products of GCRV L1
3 ÌÖÂÛ
ŚÔ1983ÄêÊŚŽÎ·ÖÀëGCRVŁŹÆùœńÒŃÓĐÊź¶àÄê”ÄÀúÊ·ĄŁÓÉÓÚGCRVșŹ11ÌőË«ÁŽ·Ö¶ÎRNA»ùÒòŚéÒÔŒ° Æä3Ąä¶ËČ»șŹpoly(A)ÎČ”ÄÌŰĐÔŁŹÊč”ĂÆä»ùÒòŚéÎÄżâ”ÄččœšŸßÓĐÒ»¶š”ÄÄŃ¶ÈŒ°žŽÔÓĐÔĄŁÔçÆÚșô łŠčÂČĄ¶ŸcDNAÎÄżâ”ÄččœšÖśÒȘœèÖúŽ«Íł”ÄcDNAșÏłÉŒ°żËÂĄ·œ·šŁŹÆäÖśÒȘŒŒÊő»ù±ŸÂ·ÏßÎȘŁșÍš čępoly(A)ŒÓÎČ·ŽÓŠ»ń”ĂŽűpoly(A)ÎČ”ÄË«ÁŽRNAŁŹŒÌ¶űČÉÓĂolig (dT)Òę”ŒcDNA”ÚÒ»ÁŽșÏłÉĄŁ žùŸĘCashdollar”ÈÈË[7]”ıš”ÀŁŹÔÚœűĐĐșôłŠčÂČĄ¶ŸąóĐÍ(strain Dearing) cDNA żËÂĄÊ±ŁŹÔűÓĂ150ŠÌgŸÍŹÎ»ËŰ(3HÄòàŚà€)±êŒÇ”ÄÈ«»ùÒòŚéRNAœűĐĐpoly(A)ŒÓÎČ·ŽÓŠ ŁŹÔÚcDNAșÏłÉʱŁŹÈÔÓĂ32P-dATPÔَαêŒÇcDNA”ÚÒ»ÁŽșÏłÉĄŁÈçŽËŽó”ÄÄŁ°ćÓĂÁż Œ°ł€Ê±Œä”Ä͏λËŰČÙŚśŁŹÔÚÒ»°ăÊ”ŃéÌőŒțÏÂÊÇÄŃÒÔœűĐĐșÍżŰÖÆ”ÄĄŁœüÄêÀŽ”ÄŚÊÁÏŒ°ÎÄÏŚÏÔÊŸ ŁŹčúÍâ¶ÔșôłŠčÂČĄ¶ŸżÆÔÓĐÁùžöÊô”Ä·ÖŚÓÉúÎïѧÌŰĐÔÒŃŃĐŸż”ĂÏà”±ÉîÈ룏ȻœöÓĐÎÄżâččœšŒ° »ùÒòŚéÆŹ¶Î”ÄÈ«ĐòÁбš”À[8ŁŹ9]ŁŹ¶űÇÒ¶ÔijЩÖŰÒȘ»ùÒòÆŹ¶Î”ÄœáččÓëčŠÄÜŒ°ŚȘÂŒ ”śżŰ·œĂæ”ÄŃĐŸżÒČÒŃÆđČœ[10ŁŹ11]ĄŁËäÈ»ÄżÇ°ŚîÎȘÆŐ±é”ÄcDNAÎÄżâččœš¶àČÉÓĂ RT-PCRșÏłÉcDNAŁŹ”«žĂ·œ·šŸùĐèœèÖúŒÆËă»úÊęŸĘżâœűĐĐÏàčŰÖÖÊôĐòÁĐ”ÄÍŹÔŽĐÔŒ°±ŁÊŰÇűÓò ”Ä·ÖÎöŁŹÒÔ»ń”ĂRT-PCR”ÄÒęÎïĐòÁĐŁŹŽÓ¶űččœšÍŹÀàČĄ¶ŸĐ¶ŸÖê”ÄcDNAÎĿ⥣ÓÉÓÚGCRVÎȘșô łŠčÂČĄ¶ŸżÆĐÂÊôĐÂÖÖŁŹÔÚÎȚ·šœűĐĐÍŹÔŽĐòÁĐ±ÈœÏ”ÄÇéżöÏÂŁŹČÉÓĂËæ»úÒęÎï·šÒę”ŒcDNAșÏłÉČ» ʧÎȘÒ»ÖÖŒò”„ĄążÉĐĐ”ÄČßÂÔĄŁÎÒĂÇÔÚÔÓĐËæ»úÒęÎï·š”Ä»ùŽĄÉÏŁŹÍščęœűĐĐһϔÁĐÌőŒț±ÈœÏŁŹ ČÉÓĂÎążËŒ¶ÄŁ°ćŁŹłÉ芔ŰččœšÁËGCRV-RNA L1ĄąL2ĄąL3ĄąM6ĄąS7ĄąS9ĄąS10ĄąS11”ÄcDNAÎÄżâ ĄŁÄżÇ°ÎÒĂÇŐęÀûÓĂËùččœš”ÄcDNAÎÄżâœűĐĐGCRVžś»ùÒòÆŹ¶Î”ÄĐòÁĐ·ÖÎöŁŹŐ✫ÎȘžĂČĄ¶ŸÒÔșó”Ä ÍŹÔŽĐÔ±ÈœÏĄąœű»ŻŃĐŸżŒ°žś»ùÒòÆŹ¶ÎËù±àÂë”Ä”°°ŚÖÊœáččÓëčŠÄÜŃĐŸż”춚ÁŒșÔĻùŽĄĄŁĄö
, °ÙĎҜҩ
»ùœđÏîÄżŁșčúŒÒŚÔÈ»żÆѧ»ùœđŚÊÖúżÎÌâ ĆúŚŒșĆŁș39570035
ŚśŐߌòœéŁș·œÇÚ(1961-)ŁŹĆźŁŹșț±±ÊĄÈËŁŹž±ŃĐŸżÔ±ĄŁÖśÒȘŽÓÊÂËźÉú¶ŻÎïČĄ¶Ÿ”Ä·ÖŚÓ ÉúÎïѧŒ°Æä»ùÒò耳ÌŃĐŸżŁŹłĐ”ŁčęčúŒÒŚÔÈ»żÆѧ»ùÒòÏîÄżŁŹ·ą±íÂÛÎÄ20ÓàÆȘĄŁ
ČÎżŒÎÄÏŚŁș
[1]żÂÀö»ȘŁŹ·œÇÚŁŹČÌÒËÈš.Ò»ÖêĐ”ÄČĘÓăłöŃȘČĄČĄ¶Ÿ·ÖÀëÎï”ÄÌŰĐÔ[J].ËźÉúÉúÎïѧ±šŁŹ1990ŁŹ14(2)Łș153Ą«159
[2]Murphy F A, Fauquet C M, Bishop D H L et al. Virus Taxnomy [J]. Arc hives of Virology, 1995,(suppl):208Ą«239
[3]ÌïŸČŁŹŚȚčđÆœŁŹ·œÇÚ.ČĘÓăłöŃȘČĄČĄ¶Ÿ»ùÒòŚé”ÄcDNAșÏłÉĄążËÂĄŒ°ĐòÁĐ·ÖÎö[J].ÖĐčúČĄ¶ŸŃ§ŁŹ1999ŁŹ14Łș87Ą«92
, http://www.100md.com
[4]·œÇÚŁŹżÂÀö»ȘŁŹČÌÒËÈš.ČĘÓăłöŃȘČĄČĄ¶Ÿ”ÄÉúł€ÌŰĐÔŒ°žß”ÎŒÛĆàŃű[J].ČĄ¶ŸŃ§ÔÓÖŸ , 1989,4(3):315Ą«319
[5]Sambrook J, Fritsch E F, Maniatis T. Molecular Cloning, A Laboratory Manu al [M], 2nd ed. USA, NY: Cold Spring Harbor, 1989.16Ą«23
[6]·œÇÚŁŹÌïŸČ.”çŚȘ»Ż·šÌážßÆœÁŹÔŰÌćDNAŚȘ»ŻĐ§ÂÊ”ÄŃĐŸż[J].ÉúÎÊőŁŹ1999ŁŹ9(4)Łș5Ą«8
[7]Cashdollar L W, Esparza, Hudson G R et al. Cloning the double-strand RNA genes of reovirus: Sequence of the cloned S2 gene [J]. Biochemistry, 1982 ,79:7644Ą«7648
, °ÙĎҜҩ
[8]Wiener J R, Joklik W K. The Sequences of the reovirus serotype 1, 2, and 3 L1 genome segments and analysis of the mode of divergence of the reovirus sero types [J]. Virology, 1989,169:194Ą«203
[9]Patton J T, Salter-CID L Kalbach A et al. Nucleotide and amino acid sequence analysis of the rotavirus non-structural RNA-binding protein NS35 [ J]. Virology, 1993,192:438Ą«446
[10]Lu G Y, Zhou Z H, Baker M L et al. Structure of double-shelle d rice dwarf virus [J].J Virol, 1998,72:8541Ą«8549
[11]Schiff L A, Nibert M L, Co M S et al. Distinct binding sites for zinc and double-stranded RNA in the reovirus outer capsid protein ŠÄ3[J]. Mol Cell Biol, 1988,8:273Ą«283
ÊŐžćÈŐÆÚŁș1999-05-21
ĐȚžćÈŐÆÚŁș1999-08-16, http://www.100md.com