œΌή’ΒΡΫαΙΙΗΡ‘λ”κœΗΑϊΕΨΜν–‘―–ΨΩ
Ής’ΏΘΚΚΈΨϋΝ÷* ’≈άώΚΆ
ΒΞΈΜΘΚ(±±Ψ©“ΫΩΤ¥σ―ßΧλ»Μ“©ΈοΦΑΖ¬…ζ“©ΈοΙζΦ“÷ΊΒψ Β―ι “Θ§ ±±Ψ© 100085)
ΙΊΦϋ¥ ΘΚœΌή’άύΥΤΈοΘΜ8-¬»œΌή’ΘΜΈ§ΦΉΥαΘΜœΗΑϊΕΨΜν–‘
“©―ß―ß±®990107.htm’Σ“Σ ΡΩΒΡΘΚ ΈΣ―Α’“–¬–ΆΩΙ÷ΉΝωΜ·ΚœΈοΚΆΧΫΧ÷Χ«ΜΖτ«ΜυΕ‘8-Cl-œΌή’ΒΡΩΙ÷ΉΝωΜν–‘ΒΡ”ΑœλΘ§Κœ≥…ΝΥœΌή’ΒΡΈ§ΦΉάύΥΤΈοΚΆ8-Cl-œΌή’―ή…ζΈοΓΘΖΫΖ®ΘΚ Ε‘œΌή’ΒΡ8ΈΜΚΆ8-Cl-œΌή’ΒΡΧ«ΜΖτ«ΜυΫχ––ΝΥΫαΙΙΗΡ‘λΘ§“‘θΘΑΖΦϋΚΆθΞΦϋ‘ΎœΌή’ΒΡ8ΈΜΝ§Ϋ”ΝΥΨΏΙ≤ινΜυΆ≈ΒΡ»βΙπθΘΜυΚΆ±ΫΦΉθΘΜυΘ§ΒΟΒΫœΌή’ΒΡΈ§ΦΉάύΥΤΈοΘΜΕ‘8-Cl-œΌή’ΒΡ5Γδ-τ«ΜυΫχ––ΝΥΦΉΜ«θΘΜ·Θ§œθΜυΜ·“‘ΦΑ¬»¥ζΖ¥”ΠΘ§ΒΟΒΫΝΥΥϋΒΡ―ή…ζΈοΓΘΫαΙϊΘΚ Κœ≥…ΝΥ(6ΓΪ12)Θ§(16)Θ§(18ΓΪ20)Β»11Ηω–¬Μ·ΚœΈοΓΘΫα¬έΘΚ ’β–©–¬Μ·ΚœΈο“‘HL-60Θ§BIUΘ§KBœΗΑϊ÷ξΒΡœΗΑϊΕΨΈΣ÷Η±ξΫχ––…ζΈοΜν–‘…Η―ΓΘ§ΫαΙϊ±μΟςΘ§8-ΈΜ»Γ¥ζΜυΚΆ5Γδ-τ«Μυ «”ΑœλœΌή’άύΜ·ΚœΈοœΗΑϊΕΨΜν–‘ΒΡ÷Ί“Σ“©–ßΜυΆ≈ΓΘ
, http://www.100md.com
STRUCTURAL MODIFICATION AND
CYTOTOXIC ACTIVITY OF ADENOSINE
He Junlin(He JL) and Zhang Lihe(Zhang LH)
(National research laboratories of natural and biomimetic drugs,Beijing Medical University, Beijing 100083)
ABSTRACT AIM: In order to find new type of antitumor compounds and study the contribution of hydroxyl groups on the sugar part to the antitumor activity of 8-Cl-adenosine, the retinoid analogs of adenosine and the derivatives of 8-Cl-adenosine were synthesized. METHODS: Based on the antitumor activity and structural feature of 8-chloroadenosine and retinoic acid, modifications were made at positions 5Γδ and 8, (un)substituted cinnamoyl and benzoyl groups were bound at 8 position through amido and ester bonds, sulfonation and nitrosation at 5Γδ-OH and its direct chlorination were conducted. RESULTS: Eleven new compounds were synthesized, they were (6ΓΪ12), (16), (18ΓΪ20). CONCLUSION: The cytotoxic activity to HL-60, BIU, and KB of the new compounds were not great. Modifications on the 5Γδ-OH and other hydroxyl groups influenced the cytotoxic activities of 8-Cl-adenosine significantly. It is sugested that the substitutions at position 8 and the hydroxyl groups on the sugar ring play an important role for the antitumor activity of adenosine analogs.
, ΑΌΡ¥“Ϋ“©
KEY WORDS adenosine analogues; 8-chloroadenosine; retinoic acid; cytotoxic activity
8-¬»œΌή’ΚΆΈ§ΦΉΥα‘ΎœΗΑϊΒΡ…ζ≥ΛΚΆΒςΫΎ÷–ΤπΉ≈Ζ«≥Θ÷Ί“ΣΒΡΉς”ΟΘ§Έ§ΦΉΥαΩ…÷ΈΝΤΑΉ―Σ≤ΓΘ§8-¬»œΌή’Ε‘»ΥΚΆΕύ÷÷≤Η»ιΕ·ΈοΒΡΕώ–‘÷ΉΝω”–“÷÷Τ…ζ≥ΛΉς”Ο[1,2]ΓΘ8-¬»œΌή’ΒΡ8ΈΜΚΆΈ§ΦΉΥαΒΡΙ≤ινΧεœΒΦΑΦΪ–‘≤ΩΖ÷Ε‘…ζΈοΜν–‘”ΑœλΦΪ¥σ[3]Θ§ΈΣ―Α’“–¬–ΆΩΙ÷ΉΝωΜ·ΚœΈοΘ§Έ“Ο«‘ΎœΌή’ΒΡ8ΈΜ“ΐ»κΖΦœψθΘΑΖΜυΘ§θΘ―θΜυΜρΚ§”–Ι≤ινΥΪΦϋΒΡΖΦœψθΘΜυΚΆθΘΑΖΜυΘ§ΡΘΡβΈ§ΦΉΥαΒΡΙ≤ιν≤ΩΖ÷Θ§άϊ”ΟΗςΉ‘ΒΡΫαΙΙΧΊΒψΫΪΝΫάύΜ·ΚœΈοΫαΚœ‘Ύ“ΜΗωΜ·ΚœΈο÷–Θ§ΜώΒΟΝΥ“Μάύ–¬–ΆΜ·ΚœΈοΓΘ
ΈΣΗΡ…Τ8-¬»œΌή’ΒΡ ΉΙΐ–ß”ΠΘ§‘ω«ΩΩΙ÷ΉΝωΉς”ΟΓΘΈ“Ο«≤…”Ο»ΐ¬»Μ·≈πΜρ»ΐδεΜ·≈πΆ―ΖΦΦΉΟ―ΒΡΦΉΜυΒΡΖΫΖ®Θ§ΫΪ8-ΦΉ―θΜυœΌή’ΒΡ8ΈΜΦΉ―θΜυΉΣΜ·ΈΣτ«ΜυΓΘ”…”ΎΖ¥”ΠΈο‘ΎΕ଻ֹΆιΚΆ¬»Ζ¬÷–ΒΡ»ήΫβΕ»–ΓΘ§Φ”»κDMF÷ζ»ή, ΖΫΖ®ΦρΒΞΘ§≤ζ¬ “≤ΗΏΒΟΕύ, ¥ο96.2%ΓΘ“‘8-Α±Μυ(τ«Μυ)-2Γδ,3Γδ,5Γδ-»ΐ““θΘΜυœΌή’ΈΣΤπ ΦΈοΚœ≥…œΌή’ΒΡΈ§ΦΉάύΥΤΈοΘ§”ΟθΘ¬»Ζ®Θ§œ»ΫΪΥα÷Τ±Η≥…θΘ¬»Θ§ ‘Ύ»ΐ““ΑΖΜρΏΝύΛΒΡ¥φ‘Ύœ¬Θ§‘Ύ “Έ¬œ¬Φ¥ΖΔ…ζΖ¥”ΠΓΘΕ‘8-¬»œΌή’ΒΡΗΡ‘λΦ·÷–‘ΎΥϋΒΡ5ΓδΈΜτ«Μυ…œΘ§ΥϋΒΡ÷±Ϋ”¬»¥ζ «Ά®Ιΐ8-¬»œΌή’”ꬻ̷―«μΩΚΆΝυΦΉΜυΝΉθΘΑΖΆξ≥…ΒΡ[4]ΓΘ”…”Ύ2ΓδΚΆ3ΓδΈΜτ«ΜυΟΜ”–±ΘΜΛΘ§Ι …ζ≥…2ΓδΚΆ3ΓδΈΜτ«Μυ±Μ―«Μ«θΘΜ·ΒΡ÷–ΦδΧε(19)Θ§ Ι÷°≤ΜΡήΖą欻¥ζΖ¥”ΠΘ§ΒΪ’β“Μ÷–ΦδΧε‘ΎΦν–‘ΧθΦΰœ¬≤ΜΈ»Ε®Θ§Β±Ζ¥”ΠΜλΚœΈο”ΟΧΦΥα«βΡΤ»ή“Κ÷–ΚΆ ±Θ§±ψΒΟΒΫ≤ζΈο(20)ΓΘ“‘2Γδ,3Γδ-‘≠ΦΉΥα““θΞΜυ-8-Cl-œΌή’(13)ΉςΈΣ÷±Ϋ”¬»¥ζΖ¥”ΠΒΡΖ¥”ΠΈοΘ§”…”Ύ±ΘΜΛΜυ‘ΎΖ¥”ΠΧθΦΰœ¬“ΉΆ―»ΞΘ§ΥυΒΟΒΡΖ¥”Π≤ζΈο «(20)ΓΘ‘ΎΫχ––5ΓδΈΜτ«ΜυΒΡθΞΜ·Ζ¥”Π«ΑΘ§œ»”Ο±ϊ≤φΜυ±ΘΜΛ8-¬»œΌή’ΒΡ2ΓδΚΆ3Γδτ«ΜυΘ§»ΜΚσ”ΟΦΉΥα»Ξ±ΘΜΛΒΟΒΫ≤ζΈοΓΘΚœ≥…¬ΖœΏΦϊΆΦ1ΓΘ
, ΑΌΡ¥“Ϋ“©
ΫΪΥυΚœ≥…ΒΡΜ·ΚœΈο“‘Ε‘HL-60Θ§ΑρκΉΑ©(BIU)ΚΆ±«― Α©(KB)œΗΑϊ÷ξΒΡœΗΑϊΕΨ–‘Ϋχ––…ζΈοΜν–‘ΒΡ≥θ≤Ϋ…Η―ΓΓΘΫαΙϊ±μΟςΘ§5Γδ-¬»-5Γδ-»Ξτ«Μυ-8-¬»œΌή’(20)Ε‘œΗΑϊΕΨΜν–‘Ήν«ΩΘ§ΫΪ’βΗωΜ·ΚœΈοΒΡ2ΓδΚΆ3ΓδΈΜτ«Μυ”Ο―«μΩΜυ±ΘΜΛΚσΘ§‘ρΟΜ”–ΝΥœΗΑϊΕΨΜν–‘ΓΘΥΒΟς8ΈΜΚΆΧ«ΜΖΒΡ3Ηωτ«ΜυΕΦΕ‘Μ·ΚœΈοΒΡ“©άμΜν–‘”–Ι±œΉΓΘ
Β―ι≤ΩΖ÷
“«Τς VG-ZAB-MS÷ ΤΉ“«Θ§300 MHz, Varian UXR-300Θ§500 MHz, Bruker AM-500ΚΥ¥≈Ι≤’ώ“«Θ§PE-240C‘ΣΥΊΖ÷Έω“«ΓΘΈόΥ°ΏΝύΛ «ΏΝύΛ(AR)Ψ≠«βΜ·ΗΤΜΊΝς24 hΚσ’τ≥ωΘ§ΈόΥ°ΦΉ¥ΦΨ≠ΦΉ¥ΦΟΨΜΊΝς8 hΚσ’τ≥ωΚσΒΟΒΫΓΘΕ଻ֹΆιΨ≠”ΟΈε―θΜ·ΕΰΝΉΜΊΝς8 hΚσ’τ≥ωΓΘ’ΙΩΣΦΝ”–“‘œ¬œΒΝ–ΘΚAΘ§¬»Ζ¬ΓΣΦΉ¥Φ(4ΓΟ1)ΘΜBΘ§““Υα““θΞΓΣ ·”ΆΟ―(3ΓΟ1)ΘΜCΘ§““Υα““θΞΓΣΜΖΦΚΆι(2ΓΟ1)ΘΜDΘ§¬»Ζ¬ΓΣΦΉ¥Φ(8ΓΟ1)ΘΜEΘ§““Υα““θΞΓΣ““¥Φ(9ΓΟ1)ΘΜFΘ§““Υα““θΞΓΣ““¥Φ(80ΓΟ1)ΓΘ
, ΑΌΡ¥“Ϋ“© 1 8-τ«ΜυœΌή’(1)
ΫΪ8-ΦΉ―θΜυœΌή’[5]–ϋΗΓ”ΎΗ…‘οΒΡCH2Cl2÷–Θ§”…”Ύ»ήΫβΕ»–ΓΘ§Φ”»κΗ…‘οΒΡDMF÷ζ»ήΘ§‘Ύ±υ―Έ‘Γά以œ¬Θ§ΒΈΦ”»ΐδεΜ·≈π(0.4 ml)ΒΡCH2Cl2»ή“Κ(5 ml)Θ§ΒΈΦ”Άξ±œΚσΘ§ΤδΦδ≥ωœ÷ΕΧ‘ίΒΡ≥Έ«εΘ§»ΜΚσ±δΜκΉ«Θ§÷±÷Ν¥σΝΩΙΧΧεΈω≥ωΓΘ “Έ¬œ¬ΦΧ–χΫΝΑη24 hΘ§“‘ ΙΖ¥”ΠΆξ»ΪΘ§ΫΪΜλΚœΈο«ψ»κ±υΥ°÷–Θ§ΫΝΑηΓΘ”–Μζ≤ψΈόΉœΆβΈϋ ’Θ§ΫΪΥ°≤ψ≈®ΥθΘ§»ΜΚσΫχ––Φθ―Ι÷υ…ΪΤΉΒΟΒΫ≤ζΈο95 mgΘ§≤ζ¬ 96.2%Θ§Rf(C) 0.38ΓΘ‘ΣΥΊΖ÷ΈωC10H13N5O5(M283)Θ§ΦΤΥψ÷Β%ΘΚC 42.40,H 4.59,N 24.73;≤βΕ®÷Β%ΘΚC 42.46,H 4.46,N 24.54ΓΘMS(FAB,M+1,284)ΓΘ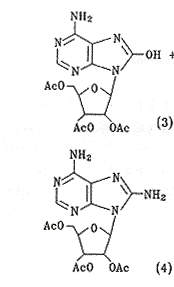







, ΑΌΡ¥“Ϋ“©
Fig 1 The synthesis of adenosine analogs.
2 8-τ«ΜυœΌή’ΒΡ““θΘΜ·Ζ¥”Π
ΫΪ8-τ«ΜυœΌή’60 mg(0.212 mmol)»ή”ΎΗ…‘οΒΡΏΝύΛ÷–Θ§œρΤδ÷–ΒΈΦ”““Υατϊ(0.23 ml)ΚσΘ§‘Ύ “Έ¬œ¬ΫΝΑη10 hΓΘTLCΦύ≤βΖ¥”ΠΈο÷ΝΆξ»Ϊ±Μ““θΘΜ·ΓΘΦθ―Ι(10 ml)’τ»Ξ¥σ≤ΩΖ÷»ήΦΝΘ§Φ”»κ±υΥ°ΫΝΑη1 hΚσΘ§”ΟCHCl3ίΆ»ΓΓΘ¥ΥCHCl3»ή“ΚΨ≠Η…‘οΘ§’τ»Ξ»ήΦΝΚσΘ§FABœ‘ Ψ≥ω”–2Ηω≤ζΈοΓΘ“ρ¥Υά©¥σΖ¥”ΠΝΩΓΘ
(1) 3.14 g(11.1 mmol)»ή”ΎΏΝύΛ150 ml÷–,‘Ύ “Έ¬œ¬ΫΝΑη,ΒΈΦ”““Υατϊ12 mlΓΘ¥ΠάμΖΫΖ®Ά§«ΑΘ§≤ζΈοΨ≠TCLœ‘ ΨΖ¥”Π«ιΩωœύΆ§ΓΘ“ρ¥Υ”ΟΦθ―Ι÷υ…ΪΤΉΖ÷άκΓΘ”ΟEtOAcΚΆ ·”ΆΟ―ΧίΕ»œ¥Ά―Θ§ΒΟΒΫ2Γδ,3Γδ,5Γδ-»ΐ““θΘΜ·≤ζΈο(3),÷Ί3.24 g,≤ζ¬ 71.4%,Rf(E) 0.22,»Ϊ““θΘΜ·≤ζΈο(5) (Φ¥8-OH“≤““θΘΜ·) 0.5 g,≤ζ¬ 10%,Rf(E) 0.70ΓΘ‘ΣΥΊΖ÷ΈωL59-1Θ§C18H29N5O9(M451)Θ§ΦΤΥψ÷Β%ΘΚC 47.89,H 4.66,N 15.52;≤βΕ®÷Β%ΘΚC 47.83,H 4.45,N 15.24ΓΘMS(FAB,M+1,452)ΓΘL59-2Θ§C16H19N5O8(M409)Θ§ΦΤΥψ÷Β%ΘΚC 46.94Θ§H 4.65Θ§N 17.11;≤βΕ®÷Β%ΘΚC 46.59Θ§H 4.35Θ§N 16.89ΓΘMS(FAB,M+1,410)ΓΘ
, ΑΌΡ¥“Ϋ“©
3 8-»βΙπθΘ―θΜυ-2Γδ,3Γδ,5Γδ-»ΐ““θΘΜυœΌή’(6)
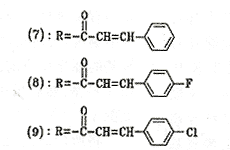
»βΙπΥα0.45 g(3 mmol)»ή”Ύ¬»Μ·―«μΩ5 ml÷–Θ§Φ”»»÷Ν90ΓφΜΊΝς4 hΚσΘ§Φθ―Ι’τ»ΞΕύ”ύΒΡ¬»Μ·―«μΩΘ§ΒΟΒΫ«≥ΜΤ…ΪΒΡ”ΆΉ¥ΈοΘ§άδ÷Ν “Έ¬ΚσΘ§Φ”»κΗ…‘οΒΡCH2Cl2 15 ml ΙΤδ»ήΫβΘ§Φ”»κ(3) 0.41 gΘ§≤ΔΒΈΦ”ΦΗΒΈΗ…‘οΒΡΏΝύΛΘ§‘Ύ “Έ¬œ¬ΫΝΑηΘ§»ή“Κ≥ ΉΊΚλ…ΪΓΘ6 hΚσΘ§TLCΦύ≤βΖ¥”ΠΈοΆξ»Ϊœϊ ßΓΘΦθ―Ι’τ»Ξ»ήΦΝΘ§”Ο≥Θ―Ι÷υ…ΪΤΉΖΫΖ®Ζ÷άκ≤ζΈοΘ§CHCl3Θ§ ·”ΆΟ―ΚΆMeOHΉςΧίΕ»œ¥Ά―ΓΘΒΟΒΫ≤ζΈο0.26 gΘ§≤ζ¬ ΈΣ42.1%Θ§Rf(D) 0.63Θ§‘ΣΥΊΖ÷ΈωC25H25N5O9(M618)Θ§ΦΤΥψ÷Β%ΘΚ C 58.04, H 4.91, N 13.22; ≤βΕ®÷Β%ΘΚ C 58.25Θ§ H 4.85Θ§ N 13.59ΓΘMS(FAB,M+1,540)ΓΘ1HNMR: ΠΡ 8.18(s,1H,2-H), 7.94(dd,2H,J=16 Hz,-CH=CH-), 7.49(m,5H,Harom), 6.12(1H,1ΓδH), 6.12(1H,2ΓδH), 5.77(m,1H,J=6.6 Hz,3ΓδH), 4.39(m,1H,J4Γδ3Γδ=6.6 Hz,4ΓδH), 4.49(dd,1H,Jab=12,Ja4Γδ=3.5 Hz,5Γδa), 4.28(dd,1H,Jb4Γδ=6.5 Hz,5Γδb), 2.12, 2.10, 2.06(s,9H,CH3CO)ΓΘ
, http://www.100md.com
4 6-»βΙπθΘΜυ-8-Εΰ»βΙπθΘΑΖΜυ-2Γδ,3Γδ,5Γδ-»ΐ““θΘΜυœΌή’(7)
(4) 0.31 g(0.76 mmol) 8-Α±Μυ-2Γδ,3Γδ,5Γδ-»ΐ““θΘΜυœΌή’[6]»ή”ΎΗ…‘οΒΡCH2Cl2»ή“Κ(10 ml)÷–Θ§ΒΈΦ”»κ»βΙπθΘ¬»0.51 g(3.04 mmol)ΒΡCH2Cl2»ή“Κ5 mlΘ§‘ΌΦ”»κΦΗΒΈ»ΐ““ΑΖΘ§‘Ύ “Έ¬œ¬ΦΧ–χΫΝΑη5 hΘ§¥ΥΜλΚœΈο”Ο≥Θ―Ι÷υ…ΪΤΉΖΫΖ®Ζ÷άκ(ΝΫ¥Έ)Θ§CHCl3Θ§ ·”ΆΟ―ΚΆMeOHΧίΕ»œ¥Ά―ΓΘΒΟΒΫ8Θ§6ΈΜ»ΐθΘΜ·≤ζΈο0.21 gΘ§≤ζ¬ ΈΣ34.7%Θ§Rf(D) 0.63Θ§‘ΣΥΊΖ÷ΈωC43H38N6O10Θ§ΦΤΥψ÷Β%ΘΚC 64.66Θ§H 4.76Θ§N 10.53;≤βΕ®÷Β%ΘΚC 65.09Θ§H 4.90Θ§N 10.26ΓΘMS(FAB,M+1,799), NMR: ΠΡ 8.71(s,1H,2-H), 7.98, 7.82, 7.75(dd,3H,JΠΝΠ¬=15.5 Hz,œ©ΜυΒΡHΠ¬), 6.77, 6.43(dd,3H,œ©ΜυHΠΝ), 6.56[s(br),1H,1ΓδH], 6.36[s(br),1H,2ΓδH], 6.13[s(br),1H,1ΓδH], 4.44(m,1H,J=3.36 Hz,5.92 Hz,4ΓδH), 4.47, 4.34(m,2H,5ΓδH), 2.18, 2.17, 2.06(s,9H,CH3CO)ΓΘ
, http://www.100md.com
5 6-(4-F-»βΙπθΘΜυ)-8-(4-F-»βΙπθΘΑΖΜυ)-2Γδ,3Γδ,5Γδ-»ΐ““θΘΜυœΌή’(8)
4-F-»βΙπΥα0.83 g(5 mmol)»ή”Ύ¬»Μ·―«μΩ10 ml÷–Θ§ΜΊΝς4 hΘ§Φθ―Ι’τ»ΞΕύ”ύΒΡ¬»Μ·―«μΩΘ§≤–”ύΈο”ΟΗ…‘οΒΡΏΝύΛ10 ml»ήΫβΘ§Φ”»κ8-Α±Μυ-2Γδ,3Γδ,5Γδ-»ΐ““θΘΜυœΌή’0.61 g(1.5 mmol)Θ§»ή“ΚΈΣΉΊΚλ…ΪΘ§ “Έ¬œ¬ΫΝΑη5 hΚσΘ§TLCΦύ≤βΖ¥”ΠΈο“―Ψ≠œϊ ßΘ§Φθ―Ι’τ»Ξ»ήΦΝΘ§ Θ”ύΈο”ΟEtOAc»ήΫβΘ§¬Υ»Ξ≤Μ»ήΈοΘ§¬Υ“Κ”Ο≥Θ―Ι÷υ…ΪΤΉΖ÷άκΘ§CHCl3Θ§ ·”ΆΟ―ΚΆMeOHΉςΧίΕ»œ¥Ά―ΓΘΒΟΒΫ8Θ§6ΈΜΕΰθΘΜ·≤ζΈο0.495 gΘ§≤ζ¬ ΈΣ46.3%Θ§Rf(D) 0.55ΓΘ‘ΣΥΊΖ÷ΈωC34H30F2N6O9.1/2H2OΘ§ΦΤΥψ÷Β%ΘΚ C 57.22Θ§ H 4.35Θ§ N 11.78; ≤βΕ®÷Β%ΘΚ C 57.20Θ§ H 4.57Θ§ N 11.52ΓΘMS(FAB,M+1,705), NMRΘΚΠΡ 8.89(s,1H,6-NH), 8.45(s,1H,2-H), 8.48,7.16(dd,2H,JΠ¬ΠΝ=15.4 Hz,Π¬-H), 6.62,6.52(dd,2H,ΠΝ-H), 7.53(8H,Harom), 7.04(8H,Harom), 7.04(1H,1ΓδH), 6.41(m,1H,2ΓδHΓδ), 6.14(brs,1H,3ΓδHΓδ), 4.44(1H,4ΓδHΓδ), 4.53(1H,5ΓδaH), 4.32(1H,5ΓδbH), 2.20(9H,CH3CO), 2.02(CH3CO)ΓΘ
, ΑΌΡ¥“Ϋ“©
6 6-(4-Cl-»βΙπθΘΜυ)-8-(4-Cl-»βΙπθΘΑΖΜυ)-2,Γδ3Γδ,5Γδ-»ΐ““θΘΜυœΌή’(9)
4-Cl-»βΙπΥα0.91 g(5 mmol)»ή”ΎΕ଻―«μΩ10 ml÷–,”Ύ90ΓφΜΊΝς5 hΚσΘ§Φθ―Ι’τ»Ξ Θ”ύΒΡΕ଻―«μΩΘ§ά以÷Ν “Έ¬Θ§”ΟΗ…‘οΒΡCH2Cl2 10 ml»ήΫβ≤–”ύΈοΘ§»ΜΚσΦ”»κ(4) 0.61 g(1.5 mmol)ΚΆ»ΐ““ΑΖ2 ml,‘Ύ “Έ¬œ¬ΫΝΑη5 hΓΘ»ή“ΚΈΣ…νΉΊΚλ…ΪΓΘΦθ―Ι’τ»Ξ»ήΦΝΘ§Φ”»κEtOAc»ήΫβΓΘΙΐ¬Υ≥ΐ»Ξ≤Μ»ήΈοΚσΘ§”Ο≥Θ―Ι÷υ…ΪΤΉΖ÷άκ(ΝΫ¥Έ)ΓΘ ·”ΆΟ―Θ§CHCl3ΧίΕ»œ¥Ά―ΓΘ”Ο““Υα““θΞΚΆ ·”ΆΟ―÷ΊΫαΨßΘ§ΒΟΒ≠ΜΤ…ΪΙΧΧε0.17 g,≤ζ¬ 15%ΓΘ Rf(D) 0.63ΓΘ‘ΣΥΊΖ÷ΈωC34H30Cl2N6O9.0.5H2O(M746), ΦΤΥψ÷Β%: C 54.69, H 4.16, N 11.26; ≤βΕ®÷Β%: C 54.70, H 4.23, N 11.48ΓΘ
, http://www.100md.com
7 6-±ΫΦΉθΘΜυ-8-±ΫΦΉθΘΑΖΜυ-2Γδ,3Γδ,5Γδ-»ΐ““θΘΜυœΌή’(10)
ΫΪΗ…‘οΒΡ8-Α±Μυ-2Γδ,3Γδ,5Γδ-»ΐ““θΘΜυœΌή’»ή”ΎΗ…‘οΒΡΏΝύΛ÷–Θ§ΒΈΦ”±ΫΦΉθΘ¬»0.21 ml,‘Ύ “Έ¬œ¬ΫΝΑη, 5 hΚσTLCΦύ≤βΖ¥”Π“―Άξ»ΪΓΘ ΫΪΜλΚœΈο’τΗ…Θ§”ΟΦθ―Ι÷υ…ΪΤΉΖ®Ζ÷άκΗςΉιΖ÷Θ§EtOAcΚΆΜΖΦΚΆιΉςΧίΕ»œ¥Ά―Θ§Rf(C) 0.72Θ§ΒΟ≤ζΈο0.27 gΘ§≤ζ¬ 60%ΓΘ‘ΣΥΊΖ÷Έω C30H27N6O9(M615)Θ§ΦΤΥψ÷Β%ΘΚ C 58.54, H 4.39, N 13.66; ≤βΕ®÷Β%ΘΚ C 58.73, H 4.57, N 13.94ΓΘ
8 6-Ε‘ΦΉ±ΫΦΉθΘΜυ-8-Ε‘ΦΉ±ΫΦΉθΘΑΖΜυ-2Γδ,3Γδ,5Γδ-»ΐ““θΘΜυ-œΌή’(11)
Ε‘ΦΉ±ΫΦΉΥα0.4 g(2.94 mmol)”ꬻ̷―«μΩ4 ml‘Ύ90ΓφΜΊΝς4 hΚσΘ§Φθ―Ι≥ΐ»ΞΙΐΝΩΒΡ¬»Μ·―«μΩΓΘΦ”»κΏΝύΛ5 ml»ήΫβ≤–ΝτΈοΓΘά以ΚσΘ§Φ”»κΒΫ8-Α±Μυ-2Γδ,3Γδ,5Γδ-»ΐ““θΘΜυœΌή’(4)0.408 g(1 mmol)ΒΡΈόΥ°ΏΝύΛ»ή“Κ(10 ml)÷–ΓΘ “Έ¬œ¬Ζ¥”Π4 hΓΘ≤ζΈο”Ο≥Θ―Ι÷υ…ΪΤΉΖ÷άκΘ§““Υα““θΞΚΆ ·”ΆΟ―ΧίΕ»œ¥Ά―ΓΘΒΟ≤ζΈο0.22 gΘ§≤ζ¬ 30.4%Θ§Rf(D) 0.57ΓΘ‘ΣΥΊΖ÷ΈωC32H32N6O9.C5H5NΘ§ΦΤΥψ÷Β%:C 61.41,H 5.12,N 13.55;≤βΕ®÷Β%:C 61.86,H 5.22,N 12.98ΓΘ1HNMR: ΠΡ 13.14(s,1H,8-NHCO), 9.04(s,1H,6-NHCO), 8.54(s,1H,2-H),7.97,7.35,7.26,8.26(m,10H,Harom), 6.62(1H,J1Γδ2Γδ=4.0 Hz,1ΓδH), 6.45(1H,J2Γδ1Γδ=4.0 Hz,J2Γδ3Γδ=6.5 Hz,2ΓδH), 5.96(1H,J3Γδ2Γδ=6.5 Hz,J3Γδ4Γδ=6.5 Hz,3ΓδH), 4.41(1H,J4Γδ3Γδ=6.5 Hz,3.5 Hz,5.5 Hz,4ΓδH), 4.51(m,1H,Jab=12.0 Hz,Ja4Γδ=3.5 Hz,5ΓδbH,5ΓδaH), 4.30(m,1H,Jba=12.0 Hz, Jb4Γδ=5.5 Hz)ΓΘ
, ΑΌΡ¥“Ϋ“©
9 6-±ΫΦΉθΘΜυ-8-±ΫΦΉθΘΑΖΜυœΌή’(12)
(10) 0.225 g(0.366 mmol)»ή”ΎCH3ONa/MeOH 4 ml(0.1 mol.L-1)»ή“ΚΚΆΗ…‘οMeOH 10 ml÷–, ‘Ύ “Έ¬œ¬Ζ¥”Π10 hΓΘ”ΟœΓ¥ΉΥα÷–ΚΆ¥ΥΜλΚœΈοΘ§»ΜΚσ”ΟΦθ―Ι÷υ…ΪΤΉΖ®Ζ÷άκΘ§ΒΟΒΫ(12) 60 mgΘ§≤ζ¬ ΈΣ33.5%ΓΘRf(D) 0.41ΓΘ‘ΣΥΊΖ÷ΈωC24H21N6O6(M489), ΦΤΥψ÷Β%: C 58.90, H 4.29, N 17.18; ≤βΕ®÷Β%: C 58.59, H 4.17, N 16.93ΓΘ1HNMR: 12.75(s,1H,8-NH), 11.75(s,1H,6-NH), 8.71(s,1H,2-H), 8.23,7.56(m,10H,Harom), 6.40(d,1H,J1Γδ2Γδ=5.5 Hz,1ΓδH), 5.20(T,1H,J2Γδ1Γδ=5.5 Hz,2ΓδH), 4.38(t,1H,J2Γδ3Γδ=5.5 Hz,J3Γδ4Γδ=9 Hz,3ΓδH), 3.98(m,1H,J4Γδ5Γδ=5.5 Hz,4ΓδH), 3.73(m,1H,Jab=12 Hz,Ja4Γδ=4.5 Hz,5ΓδaH), 3.56(m,1H,JbΓδ4Γδ=5.5 Hz,5ΓδbH)ΓΘ
, ΑΌΡ¥“Ϋ“©
10 2Γδ,3Γδ-‘≠ΦΉΥα““θΞΜυ-8-Cl-œΌή’(13)
8-Cl-œΌή’2 g(6.62 mmol)ΚΆ‘≠ΦΉΥα““θΞ5.3 mlΘΜ»ΐ¬»““Υα4.66 gΚΆΕΰ―θΝυΜΖ53 mlΒΡΜλΚœΈο‘Ύ60ΓφΦ”»»Ζ¥”Π4 hΘ§TLCΦύ≤βΖ¥”ΠΆξ»Ϊ[7]ΓΘ”ΟCH2Cl2 100 mlœΓ Ά¥Υ»ή“ΚΘ§»ΜΚσ”ΟNaCO3»ή“ΚΚΆH2OΗςœ¥3¥ΈΓΘΗ…‘οΘ§≥ΐ»Ξ»ήΦΝΚσΘ§”ΟΈόΥ°EtOH÷ΊΫαΨßΘ§ΒΟΒΫ≤ζΈο2.11 gΘ§≤ζ¬ 89.2%ΓΘNMR÷Λ ΒΈΣΖ«Ε‘”≥“λΙΙΧεΜλΚœΈοΘ§ Rf(D) 0.87ΓΘ1HNMR: “λΙΙΧεI,ΠΡ 8.19(s,1H,2-H), 7.57(s,2H,6-NH2), 6.23(s,1H,‘≠ΦΉΥα““θΞΒΡH), 6.20(d,1H,J1Γδ2Γδ=2.7 Hz,1ΓδH), 5.78(dd,1H,J1Γδ2Γδ=2.7 Hz,J2Γδ3Γδ=6.6 Hz,2ΓδH), 5.14(m,1H,J3Γδ4Γδ=3.6 Hz,3ΓδH), 4.28(m,1H,J4Γδ5Γδ=6.6 Hz,4ΓδH), 3.50ΓΪ3.37(m,2H,5ΓδH), 4.21(m,2H,J=6.6 Hz,-CH2-), 1.22(t,3H,-CH3), 5.16ΓΪ5.01(br,1H,5Γδ-OH)ΓΘ“λΙΙΧεIIΘ§ ΠΡ 8.17(s,1H,2-H), 7.57(s,2H,6-NH2), 6.17(s,1H,‘≠ΦΉΥαθΞΒΡH), 6.07(s,1H,1ΓδH), 5.73(dd,1H,J1Γδ2Γδ=3 Hz,2ΓδH), 5.02(m,1H,J3Γδ4Γδ=3.6 Hz,3ΓδH), 4.19(m,1H,J4Γδ5Γδ=3.8 Hz,4ΓδH), 3.50ΓΪ3.37(m,1H,5ΓδH), 4.21(m,2H,J=6.6,-CH2-), 1.13(t,3H,CH3), 5.16ΓΪ5.01(m,1H,5Γδ-OH)ΓΘ
, ΑΌΡ¥“Ϋ“©
11 2Γδ,3Γδ-±ϊ≤φΜυ-8-Cl-œΌή’(14)
8-Cl-œΌή’0.3 g(1 mmol)”ΎΤ’Ά®±ϊΆΣ÷–ΨγΝ“ΫΝΑηΘ§‘Ύ±υ‘Γά以œ¬Θ§Ζ÷≈ζΒΈΦ”ΝρΥα0.15 mlΓΘΈ§≥÷Ζ¥”ΠΈ¬Ε»‘Ύ(0.5ΓΪ10)ΓφΓΘΒΈΦ”Άξ±œΚσΘ§‘ΌΦΧ–χΫΝΑη5 hΓΘ‘Ύ¥ΥΤΎΦδΘ§Έ¬Ε»÷πΫΞ…ΐ÷Ν “Έ¬ΓΘΖ¥”ΠΆξ≥…ΚσΘ§‘Ύ±υ‘Γά以œ¬Θ§”ΟNaOH»ή“Κ÷–ΚΆ÷Ν÷––‘Θ§Ιΐ“ΙΘ§Ιΐ¬Υ≥ΐ»Ξ―ΈΘ§¬Υ“Κ≈®ΥθΚσ”Ο¬»Ζ¬»ήΫβΘ§Υ°œ¥¥Υ¬»Ζ¬»ή“ΚΘ§≤Δ”Ο≥Θ―Ι÷υ…ΪΤΉΖ®Ζ÷άκ≥ω≤ζΈο0.32 g, Rf(C) 0.89,‘ΣΥΊΖ÷ΈωC13H16ClN5O4(M342)Θ§ΦΤΥψ÷Β%: C 45.61, H 4.68, N 20.47; ≤βΕ®÷Β%: C 45.24, H 4.47, N 20.24ΓΘ
12 2Γδ,3Γδ-±ϊ≤φΜυ-5Γδ-O-ΦΉΜ«θΘΜυ-8-Cl-œΌή’(15)[8]
, ΑΌΡ¥“Ϋ“©
ΫΪ(14) 1 g(2.93 mmol)»ή”ΎΗ…‘οΏΝύΛ30 ml÷–Θ§‘Ύ±υ‘Γά以œ¬Θ§ΒΈΦ”ΦΉΜ«θΘ¬»0.5 mlΓΘ»ή“ΚΦ¥±δΜΤ…ΪΓΘ‘Ύ¥ΥΈ¬Ε»œ¬ΫΝΑη4 hΓΘΦθ―Ι’τ»Ξ»ήΦΝΘ§”ΟΦθ―Ι÷υ…ΪΤΉΖ®Ζ÷άκΓΘEtOAcΚΆEtOHΧίΕ»œ¥Ά―Θ§ΒΟ≤ζΈο0.39 gΘ§≤ζ¬ 31.7%Θ§Rf(B) 0.78ΓΘ
13 5Γδ-O-ΦΉΜ«θΘΜυ-8-Cl-œΌή’(16)
ΫΪ(15) 0.21 g(0.5 mmol)»ή”ΎHCOOH(80%) 2.5 ml÷–Θ§ “Έ¬œ¬ΫΝΑη10 hΓΘ»ΜΚσΦθ―Ι’τ»ΞHCOOHΘ§≤Δ”ΟΈόΥ°EtOH÷Ί’τ3¥ΈΓΘ≤–”ύΈο”Ο≥Θ―Ι÷υ…ΪΤΉΖ®Ζ÷άκΓΘ ·”ΆΟ―ΚΆ““Υα““θΞΉςΧίΕ»œ¥Ά―Θ§ΒΟΒΫΑΉ…ΪΙΧΧε0.13 g,≤ζ¬ 68%Θ§‘ΣΥΊΖ÷ΈωC11H14ClN5O6S(M380),ΦΤΥψ÷Β%:C 34.74,H 3.68,N 18.42;≤βΕ®÷Β%:C 34.97,H 3.72,N 18.19ΓΘ
, ΑΌΡ¥“Ϋ“©
14 2Γδ,3Γδ-±ϊ≤φΜυ-5Γδ-O-œθΜυ-8-Cl-œΌή’(17)
ΫΪ≈®HNO3 1.4 mlΒΈΦ”»κ±υ―Έ‘Γά以ΒΡΒ»ΧεΜΐΒΡ““Υατϊ÷–Θ§ΒΟΒΫΜλΚœΥατϊΓΘ(14) 0.26 g(0.7 mmol) »ή”ΎEtOAc÷–Θ§”Ο±υ―Έ‘Γά以÷Ν-30ΓφΓΘ»ΓΜλΚœΥατϊ1 mlΒΈΦ”»κ(14)ΒΡ»ή“Κ÷–Θ§Ζ¥”ΠΜλΚœΈο‘Ύ¥ΥΈ¬Ε»œ¬ΦΧ–χΖ¥”ΠΘ§4.5 hΚσΘ§”ΟNaHCO3»ή“Κ÷–ΚΆΓΘ”–Μζ≤ψΚΆH2O≤ψΖ÷ΩΣΘ§H2O≤ψ”ΟEtOAcίΆ»Γ3¥ΈΓΘΚœ≤ΔEtOAc»ή“ΚΘ§Η…‘οΘ§”ΟΦθ―Ι÷υ…ΪΤΉΖ®ΒΟΒΫ0.2 g≤ζΈο(ΑΉ…ΪΙΧΧε)Θ§≤ζ¬ 74.1%,Rf(C) 0.44ΓΘ‘ΣΥΊΖ÷ΈωC13H15ClN6O6.H2O(M387),ΦΤΥψ÷Β%ΘΚ C 40.31, H 3.88, N 21.71ΘΜ ≤βΕ®÷Β%ΘΚ C 39.84, H 3.96, N 21.46ΓΘ
, ΑΌΡ¥“Ϋ“©
15 (17)ΒΡ»Ξ±ΘΜΛΖ¥”Π5Γδ-O-œθΜυ-8-Cl-œΌή’(18)
ΫΪ(17)»ή”Ύ88%ΒΡHCOOH÷–Θ§‘Ύ “Έ¬œ¬ΫΝΑη10 hΘ§Φθ―Ι’τ»ΞHCOOHΘ§‘Ό”ΟΈόΥ°EtOH÷Ί’τ3¥ΈΓΘΖ¥”ΠΜλΚœΈο”Ο≥Θ―Ι÷υ…ΪΤΉΖ®Ζ÷άκΓΘEtOAcΚΆ ·”ΆΟ―ΧίΕ»œ¥Ά―ΓΘΒΟΒΫ≤ζΈο60 mgΘ§≤ζ¬ 61.8%ΓΘRf(B) 0.74Θ§‘ΣΥΊΖ÷ΈωC10H11ClN6O6(M347), ΦΤΥψ÷Β%ΘΚ C 34.58, H 3.17, N 24.21ΘΜ ≤βΕ®÷Β%ΘΚ C 34.83, H 3.32, N 23.86ΓΘ
16 5Γδ-¬»-5Γδ-»Ξ―θ-2Γδ,3Γδ-―«μΩΜυ-8-Cl-œΌή’(19)
ΫΪ(13)¥ζΧφ8-Cl-œΌή’”κHMPAΚΆSOCl2ΒΡΜλΚœΈοΖ¥”ΠΓΘΖ¥”ΠΜλΚœΈο”ΟΦθ―Ι÷υ…ΪΤΉΖ÷άκΘ§““Υα““θΞΓΔΜΖΦΚΆιΚΆEtOHΧίΕ»œ¥Ά―,ΒΟΒΫ≤ζΈο0.2 gΘ§≤ζ¬ 32.8%ΓΘRf(F) 0.78ΓΘ‘ΣΥΊΖ÷ΈωC10H9Cl2N5O4S.H2O(M370.5), ΦΤΥψ÷Β%ΘΚ C 32.38, H 2.56, N 18.89ΘΜ ≤βΕ®÷Β%ΘΚ C 32.78, H 2.53, N 18.57ΓΘ1HNMRΘΚ ΠΡ 6.43(d,1H,J1Γδ2Γδ=2.4 Hz,1ΓδH), 6.39(m,1H,J1Γδ2Γδ=2.4 Hz,J2Γδ,3Γδ=7.5 Hz,2ΓδH), 5.87(m,1H,J2Γδ,3Γδ=7.5 Hz,J3Γδ,4Γδ=3.9 Hz,3ΓδH), 4.73(m,1H,J=3.9,6.0,7.2 Hz,4ΓδH), 3.86(m,2H,Jab=11.1, Ja4Γδ=7.2 Hz,Jb4Γδ=6.3 Hz,5ΓδH), 7.64(s,2H,6-NH2), 8.19(s,1H,2-H)ΓΘ
, ΑΌΡ¥“Ϋ“©
17 5Γδ-¬»-5Γδ-»Ξ―θ-8-Cl-œΌή’(20)[9]
ΝυΦΉΜυΝΉθΘΑΖ(HMPA)1.6 ml”Ο±υ‘Γά以ȧœρΤδ÷–ΒΈΦ”SOCl2 0.24 mlΓΘ‘Ύ0ΓφΫΝΑηœ¬15 minΚσΘ§ΆΕ»κ8-Cl-œΌή’0.3 g(1 mmol)ΓΘΖ¥”Π2 hΓΘ “Έ¬œ¬ΫΝΑηΙΐ“ΙΓΘ»ή“Κ≥ œ Κλ…ΪΓΘΫΪΜλΚœΈο«ψ»κ±υH2O 25 ml÷–Θ§NaHCO3»ή“Κ÷–ΚΆ÷Ν÷––‘Θ§”ΟΦθ―Ι÷υ…ΪΤΉΖ÷άκΘ§EtOAcΚΆEtOHΧίΕ»œ¥Ά―ΓΘΒΟΒΫΑΉ…ΪΙΧΧε0.1 gΘ§≤ζ¬ 31.3%,Rf(B) 0.90ΓΘ‘ΣΥΊΖ÷ΈωC10H11Cl2N5O3Θ§ΦΤΥψ÷Β%ΘΚ C 37.50, H 3.44, N 21.88ΘΜ ≤βΕ®÷Β%ΘΚ C 37.87, H 3.78, N 21.48ΓΘ
÷¬–Μ Άθ ι”ώΫΧ ΎΧαΙ©»Γ¥ζ»βΙπΥαΘΜ÷ΊΒψ Β―ι “ΒΡ‘ΣΥΊΖ÷Έω Β―ι “ΚΆΚΥ¥≈ “Θ§÷––Ρ Β―ι “÷ ΤΉ “Θ§±±Ψ©¥σ―ßΚΆΖάΜ·‘ΚΚΥ¥≈ “Θ§÷–ΙζΩΤ―ß‘ΚΜ·―ßΥυ–≠ΉςΆξ≥…ΝΥΚΥ¥≈Ι≤’ώΘ§÷ ΤΉΚΆ‘ΣΥΊΖ÷ΈωΘΜ÷ΊΒψ Β―ι “Άθ»π«δΒ»άœ ΠΆξ≥…≥θ≤Ϋ…ζΈοΜν–‘…Η―Γ Β―ιΓΘ
, ΑΌΡ¥“Ϋ“©
*ΝΣœΒ»Υœ÷÷ΖΘΚΨϋ ¬“Ϋ―ßΩΤ―ß‘ΚΕΨΈο“©Έο―–ΨΩΥυΘ§±±Ψ© 100850
Tel:(010)66887429-66611
≤ΈΩΦΈΡœΉ
1 ΖΫΦ“¥ΜΘ§ ·”άΫχΘ§≈μΟτΘ§Β». 8-¬»œΌή’ΒΡΩΙ÷ΉΝωΉς”Ο―–ΨΩ. ÷–ΜΣ÷ΉΝω‘”÷Ψ, 1995,17ΓΟ5
2 ΆθœήλβΘ§÷ΘΒ¬œ»Θ§ΝθνΘΘ§Β». 8-¬»œΌή’”’ΒΦMOLT-4œΗΑϊΒρΆω. ΩΤ―ßΆ®±®, 1997,42ΓΟ189
3 Lin Z, Nadzan AM, Heyman RA, et al. Discovery of novel retinoicacid receptor agonists having potent antiproliferative activity in cervical cancer cells. J Med Chem, 1996,39ΓΟ2659
, ΑΌΡ¥“Ϋ“©
4 Holy A, Rosenberg I. An improved synthesis of S-adenosyl-L-nomocystein and related compounds. Collect Czech Chem Commun, 1985,50ΓΟ1514
5 Long RA, Robins RK, Townsend LB. Purine nucleosides, XV. The synthesis of 8-amino- and 8-substituted aminopurine nucleosides. Biochemistry, 1967,32ΓΟ2751
6 Ikehara M, Yamada S. Studies of nucleosides and nucleotides. XLIX. Synthesis of 8-fluoroadenosine. Chem Pharm Bull, 1971,19ΓΟ104
, http://www.100md.com
7 Eckstein F, Cramer F. Notize uber 2Γδ,3Γδ-O-athoxy-methylen-derivate von Ribonucleosiden. Chem Ber, 1965ΓΟ995
8 Sharma M, Wikiel H, Hromchak R, et al. Synthesis of 5Γδ-substituted derivatives of the pyrrolo[2,3-d]-pyrimidine nucleoside sangivamycin and their effect on protein kinase A and C. Nucleosides and Nudeotides, 1993,12(3 & 4)ΓΟ295
9 Prisbe EJ, Smejkal J, Verheyden JPH, et al. Halo sugar nucleosides. 5. Synthesis of angustmycin A and some base analogues. J Org Chem, 1976,41ΓΟ1836
’Ηε»’ΤΎ: 1997-11-28, ΑΌΡ¥“Ϋ“©(ΚΈΨϋΝ÷* ’≈άώΚΆ)
ΒΞΈΜΘΚ(±±Ψ©“ΫΩΤ¥σ―ßΧλ»Μ“©ΈοΦΑΖ¬…ζ“©ΈοΙζΦ“÷ΊΒψ Β―ι “Θ§ ±±Ψ© 100085)
ΙΊΦϋ¥ ΘΚœΌή’άύΥΤΈοΘΜ8-¬»œΌή’ΘΜΈ§ΦΉΥαΘΜœΗΑϊΕΨΜν–‘
“©―ß―ß±®990107.htm’Σ“Σ ΡΩΒΡΘΚ ΈΣ―Α’“–¬–ΆΩΙ÷ΉΝωΜ·ΚœΈοΚΆΧΫΧ÷Χ«ΜΖτ«ΜυΕ‘8-Cl-œΌή’ΒΡΩΙ÷ΉΝωΜν–‘ΒΡ”ΑœλΘ§Κœ≥…ΝΥœΌή’ΒΡΈ§ΦΉάύΥΤΈοΚΆ8-Cl-œΌή’―ή…ζΈοΓΘΖΫΖ®ΘΚ Ε‘œΌή’ΒΡ8ΈΜΚΆ8-Cl-œΌή’ΒΡΧ«ΜΖτ«ΜυΫχ––ΝΥΫαΙΙΗΡ‘λΘ§“‘θΘΑΖΦϋΚΆθΞΦϋ‘ΎœΌή’ΒΡ8ΈΜΝ§Ϋ”ΝΥΨΏΙ≤ινΜυΆ≈ΒΡ»βΙπθΘΜυΚΆ±ΫΦΉθΘΜυΘ§ΒΟΒΫœΌή’ΒΡΈ§ΦΉάύΥΤΈοΘΜΕ‘8-Cl-œΌή’ΒΡ5Γδ-τ«ΜυΫχ––ΝΥΦΉΜ«θΘΜ·Θ§œθΜυΜ·“‘ΦΑ¬»¥ζΖ¥”ΠΘ§ΒΟΒΫΝΥΥϋΒΡ―ή…ζΈοΓΘΫαΙϊΘΚ Κœ≥…ΝΥ(6ΓΪ12)Θ§(16)Θ§(18ΓΪ20)Β»11Ηω–¬Μ·ΚœΈοΓΘΫα¬έΘΚ ’β–©–¬Μ·ΚœΈο“‘HL-60Θ§BIUΘ§KBœΗΑϊ÷ξΒΡœΗΑϊΕΨΈΣ÷Η±ξΫχ––…ζΈοΜν–‘…Η―ΓΘ§ΫαΙϊ±μΟςΘ§8-ΈΜ»Γ¥ζΜυΚΆ5Γδ-τ«Μυ «”ΑœλœΌή’άύΜ·ΚœΈοœΗΑϊΕΨΜν–‘ΒΡ÷Ί“Σ“©–ßΜυΆ≈ΓΘ
, http://www.100md.com
STRUCTURAL MODIFICATION AND
CYTOTOXIC ACTIVITY OF ADENOSINE
He Junlin(He JL) and Zhang Lihe(Zhang LH)
(National research laboratories of natural and biomimetic drugs,Beijing Medical University, Beijing 100083)
ABSTRACT AIM: In order to find new type of antitumor compounds and study the contribution of hydroxyl groups on the sugar part to the antitumor activity of 8-Cl-adenosine, the retinoid analogs of adenosine and the derivatives of 8-Cl-adenosine were synthesized. METHODS: Based on the antitumor activity and structural feature of 8-chloroadenosine and retinoic acid, modifications were made at positions 5Γδ and 8, (un)substituted cinnamoyl and benzoyl groups were bound at 8 position through amido and ester bonds, sulfonation and nitrosation at 5Γδ-OH and its direct chlorination were conducted. RESULTS: Eleven new compounds were synthesized, they were (6ΓΪ12), (16), (18ΓΪ20). CONCLUSION: The cytotoxic activity to HL-60, BIU, and KB of the new compounds were not great. Modifications on the 5Γδ-OH and other hydroxyl groups influenced the cytotoxic activities of 8-Cl-adenosine significantly. It is sugested that the substitutions at position 8 and the hydroxyl groups on the sugar ring play an important role for the antitumor activity of adenosine analogs.
, ΑΌΡ¥“Ϋ“©
KEY WORDS adenosine analogues; 8-chloroadenosine; retinoic acid; cytotoxic activity
8-¬»œΌή’ΚΆΈ§ΦΉΥα‘ΎœΗΑϊΒΡ…ζ≥ΛΚΆΒςΫΎ÷–ΤπΉ≈Ζ«≥Θ÷Ί“ΣΒΡΉς”ΟΘ§Έ§ΦΉΥαΩ…÷ΈΝΤΑΉ―Σ≤ΓΘ§8-¬»œΌή’Ε‘»ΥΚΆΕύ÷÷≤Η»ιΕ·ΈοΒΡΕώ–‘÷ΉΝω”–“÷÷Τ…ζ≥ΛΉς”Ο[1,2]ΓΘ8-¬»œΌή’ΒΡ8ΈΜΚΆΈ§ΦΉΥαΒΡΙ≤ινΧεœΒΦΑΦΪ–‘≤ΩΖ÷Ε‘…ζΈοΜν–‘”ΑœλΦΪ¥σ[3]Θ§ΈΣ―Α’“–¬–ΆΩΙ÷ΉΝωΜ·ΚœΈοΘ§Έ“Ο«‘ΎœΌή’ΒΡ8ΈΜ“ΐ»κΖΦœψθΘΑΖΜυΘ§θΘ―θΜυΜρΚ§”–Ι≤ινΥΪΦϋΒΡΖΦœψθΘΜυΚΆθΘΑΖΜυΘ§ΡΘΡβΈ§ΦΉΥαΒΡΙ≤ιν≤ΩΖ÷Θ§άϊ”ΟΗςΉ‘ΒΡΫαΙΙΧΊΒψΫΪΝΫάύΜ·ΚœΈοΫαΚœ‘Ύ“ΜΗωΜ·ΚœΈο÷–Θ§ΜώΒΟΝΥ“Μάύ–¬–ΆΜ·ΚœΈοΓΘ
ΈΣΗΡ…Τ8-¬»œΌή’ΒΡ ΉΙΐ–ß”ΠΘ§‘ω«ΩΩΙ÷ΉΝωΉς”ΟΓΘΈ“Ο«≤…”Ο»ΐ¬»Μ·≈πΜρ»ΐδεΜ·≈πΆ―ΖΦΦΉΟ―ΒΡΦΉΜυΒΡΖΫΖ®Θ§ΫΪ8-ΦΉ―θΜυœΌή’ΒΡ8ΈΜΦΉ―θΜυΉΣΜ·ΈΣτ«ΜυΓΘ”…”ΎΖ¥”ΠΈο‘ΎΕ଻ֹΆιΚΆ¬»Ζ¬÷–ΒΡ»ήΫβΕ»–ΓΘ§Φ”»κDMF÷ζ»ή, ΖΫΖ®ΦρΒΞΘ§≤ζ¬ “≤ΗΏΒΟΕύ, ¥ο96.2%ΓΘ“‘8-Α±Μυ(τ«Μυ)-2Γδ,3Γδ,5Γδ-»ΐ““θΘΜυœΌή’ΈΣΤπ ΦΈοΚœ≥…œΌή’ΒΡΈ§ΦΉάύΥΤΈοΘ§”ΟθΘ¬»Ζ®Θ§œ»ΫΪΥα÷Τ±Η≥…θΘ¬»Θ§ ‘Ύ»ΐ““ΑΖΜρΏΝύΛΒΡ¥φ‘Ύœ¬Θ§‘Ύ “Έ¬œ¬Φ¥ΖΔ…ζΖ¥”ΠΓΘΕ‘8-¬»œΌή’ΒΡΗΡ‘λΦ·÷–‘ΎΥϋΒΡ5ΓδΈΜτ«Μυ…œΘ§ΥϋΒΡ÷±Ϋ”¬»¥ζ «Ά®Ιΐ8-¬»œΌή’”ꬻ̷―«μΩΚΆΝυΦΉΜυΝΉθΘΑΖΆξ≥…ΒΡ[4]ΓΘ”…”Ύ2ΓδΚΆ3ΓδΈΜτ«ΜυΟΜ”–±ΘΜΛΘ§Ι …ζ≥…2ΓδΚΆ3ΓδΈΜτ«Μυ±Μ―«Μ«θΘΜ·ΒΡ÷–ΦδΧε(19)Θ§ Ι÷°≤ΜΡήΖą欻¥ζΖ¥”ΠΘ§ΒΪ’β“Μ÷–ΦδΧε‘ΎΦν–‘ΧθΦΰœ¬≤ΜΈ»Ε®Θ§Β±Ζ¥”ΠΜλΚœΈο”ΟΧΦΥα«βΡΤ»ή“Κ÷–ΚΆ ±Θ§±ψΒΟΒΫ≤ζΈο(20)ΓΘ“‘2Γδ,3Γδ-‘≠ΦΉΥα““θΞΜυ-8-Cl-œΌή’(13)ΉςΈΣ÷±Ϋ”¬»¥ζΖ¥”ΠΒΡΖ¥”ΠΈοΘ§”…”Ύ±ΘΜΛΜυ‘ΎΖ¥”ΠΧθΦΰœ¬“ΉΆ―»ΞΘ§ΥυΒΟΒΡΖ¥”Π≤ζΈο «(20)ΓΘ‘ΎΫχ––5ΓδΈΜτ«ΜυΒΡθΞΜ·Ζ¥”Π«ΑΘ§œ»”Ο±ϊ≤φΜυ±ΘΜΛ8-¬»œΌή’ΒΡ2ΓδΚΆ3Γδτ«ΜυΘ§»ΜΚσ”ΟΦΉΥα»Ξ±ΘΜΛΒΟΒΫ≤ζΈοΓΘΚœ≥…¬ΖœΏΦϊΆΦ1ΓΘ
, ΑΌΡ¥“Ϋ“©
ΫΪΥυΚœ≥…ΒΡΜ·ΚœΈο“‘Ε‘HL-60Θ§ΑρκΉΑ©(BIU)ΚΆ±«― Α©(KB)œΗΑϊ÷ξΒΡœΗΑϊΕΨ–‘Ϋχ––…ζΈοΜν–‘ΒΡ≥θ≤Ϋ…Η―ΓΓΘΫαΙϊ±μΟςΘ§5Γδ-¬»-5Γδ-»Ξτ«Μυ-8-¬»œΌή’(20)Ε‘œΗΑϊΕΨΜν–‘Ήν«ΩΘ§ΫΪ’βΗωΜ·ΚœΈοΒΡ2ΓδΚΆ3ΓδΈΜτ«Μυ”Ο―«μΩΜυ±ΘΜΛΚσΘ§‘ρΟΜ”–ΝΥœΗΑϊΕΨΜν–‘ΓΘΥΒΟς8ΈΜΚΆΧ«ΜΖΒΡ3Ηωτ«ΜυΕΦΕ‘Μ·ΚœΈοΒΡ“©άμΜν–‘”–Ι±œΉΓΘ
Β―ι≤ΩΖ÷
“«Τς VG-ZAB-MS÷ ΤΉ“«Θ§300 MHz, Varian UXR-300Θ§500 MHz, Bruker AM-500ΚΥ¥≈Ι≤’ώ“«Θ§PE-240C‘ΣΥΊΖ÷Έω“«ΓΘΈόΥ°ΏΝύΛ «ΏΝύΛ(AR)Ψ≠«βΜ·ΗΤΜΊΝς24 hΚσ’τ≥ωΘ§ΈόΥ°ΦΉ¥ΦΨ≠ΦΉ¥ΦΟΨΜΊΝς8 hΚσ’τ≥ωΚσΒΟΒΫΓΘΕ଻ֹΆιΨ≠”ΟΈε―θΜ·ΕΰΝΉΜΊΝς8 hΚσ’τ≥ωΓΘ’ΙΩΣΦΝ”–“‘œ¬œΒΝ–ΘΚAΘ§¬»Ζ¬ΓΣΦΉ¥Φ(4ΓΟ1)ΘΜBΘ§““Υα““θΞΓΣ ·”ΆΟ―(3ΓΟ1)ΘΜCΘ§““Υα““θΞΓΣΜΖΦΚΆι(2ΓΟ1)ΘΜDΘ§¬»Ζ¬ΓΣΦΉ¥Φ(8ΓΟ1)ΘΜEΘ§““Υα““θΞΓΣ““¥Φ(9ΓΟ1)ΘΜFΘ§““Υα““θΞΓΣ““¥Φ(80ΓΟ1)ΓΘ
, ΑΌΡ¥“Ϋ“© 1 8-τ«ΜυœΌή’(1)
ΫΪ8-ΦΉ―θΜυœΌή’[5]–ϋΗΓ”ΎΗ…‘οΒΡCH2Cl2÷–Θ§”…”Ύ»ήΫβΕ»–ΓΘ§Φ”»κΗ…‘οΒΡDMF÷ζ»ήΘ§‘Ύ±υ―Έ‘Γά以œ¬Θ§ΒΈΦ”»ΐδεΜ·≈π(0.4 ml)ΒΡCH2Cl2»ή“Κ(5 ml)Θ§ΒΈΦ”Άξ±œΚσΘ§ΤδΦδ≥ωœ÷ΕΧ‘ίΒΡ≥Έ«εΘ§»ΜΚσ±δΜκΉ«Θ§÷±÷Ν¥σΝΩΙΧΧεΈω≥ωΓΘ “Έ¬œ¬ΦΧ–χΫΝΑη24 hΘ§“‘ ΙΖ¥”ΠΆξ»ΪΘ§ΫΪΜλΚœΈο«ψ»κ±υΥ°÷–Θ§ΫΝΑηΓΘ”–Μζ≤ψΈόΉœΆβΈϋ ’Θ§ΫΪΥ°≤ψ≈®ΥθΘ§»ΜΚσΫχ––Φθ―Ι÷υ…ΪΤΉΒΟΒΫ≤ζΈο95 mgΘ§≤ζ¬ 96.2%Θ§Rf(C) 0.38ΓΘ‘ΣΥΊΖ÷ΈωC10H13N5O5(M283)Θ§ΦΤΥψ÷Β%ΘΚC 42.40,H 4.59,N 24.73;≤βΕ®÷Β%ΘΚC 42.46,H 4.46,N 24.54ΓΘMS(FAB,M+1,284)ΓΘ
, ΑΌΡ¥“Ϋ“©
Fig 1 The synthesis of adenosine analogs.
2 8-τ«ΜυœΌή’ΒΡ““θΘΜ·Ζ¥”Π
ΫΪ8-τ«ΜυœΌή’60 mg(0.212 mmol)»ή”ΎΗ…‘οΒΡΏΝύΛ÷–Θ§œρΤδ÷–ΒΈΦ”““Υατϊ(0.23 ml)ΚσΘ§‘Ύ “Έ¬œ¬ΫΝΑη10 hΓΘTLCΦύ≤βΖ¥”ΠΈο÷ΝΆξ»Ϊ±Μ““θΘΜ·ΓΘΦθ―Ι(10 ml)’τ»Ξ¥σ≤ΩΖ÷»ήΦΝΘ§Φ”»κ±υΥ°ΫΝΑη1 hΚσΘ§”ΟCHCl3ίΆ»ΓΓΘ¥ΥCHCl3»ή“ΚΨ≠Η…‘οΘ§’τ»Ξ»ήΦΝΚσΘ§FABœ‘ Ψ≥ω”–2Ηω≤ζΈοΓΘ“ρ¥Υά©¥σΖ¥”ΠΝΩΓΘ
(1) 3.14 g(11.1 mmol)»ή”ΎΏΝύΛ150 ml÷–,‘Ύ “Έ¬œ¬ΫΝΑη,ΒΈΦ”““Υατϊ12 mlΓΘ¥ΠάμΖΫΖ®Ά§«ΑΘ§≤ζΈοΨ≠TCLœ‘ ΨΖ¥”Π«ιΩωœύΆ§ΓΘ“ρ¥Υ”ΟΦθ―Ι÷υ…ΪΤΉΖ÷άκΓΘ”ΟEtOAcΚΆ ·”ΆΟ―ΧίΕ»œ¥Ά―Θ§ΒΟΒΫ2Γδ,3Γδ,5Γδ-»ΐ““θΘΜ·≤ζΈο(3),÷Ί3.24 g,≤ζ¬ 71.4%,Rf(E) 0.22,»Ϊ““θΘΜ·≤ζΈο(5) (Φ¥8-OH“≤““θΘΜ·) 0.5 g,≤ζ¬ 10%,Rf(E) 0.70ΓΘ‘ΣΥΊΖ÷ΈωL59-1Θ§C18H29N5O9(M451)Θ§ΦΤΥψ÷Β%ΘΚC 47.89,H 4.66,N 15.52;≤βΕ®÷Β%ΘΚC 47.83,H 4.45,N 15.24ΓΘMS(FAB,M+1,452)ΓΘL59-2Θ§C16H19N5O8(M409)Θ§ΦΤΥψ÷Β%ΘΚC 46.94Θ§H 4.65Θ§N 17.11;≤βΕ®÷Β%ΘΚC 46.59Θ§H 4.35Θ§N 16.89ΓΘMS(FAB,M+1,410)ΓΘ
, ΑΌΡ¥“Ϋ“©
3 8-»βΙπθΘ―θΜυ-2Γδ,3Γδ,5Γδ-»ΐ““θΘΜυœΌή’(6)
»βΙπΥα0.45 g(3 mmol)»ή”Ύ¬»Μ·―«μΩ5 ml÷–Θ§Φ”»»÷Ν90ΓφΜΊΝς4 hΚσΘ§Φθ―Ι’τ»ΞΕύ”ύΒΡ¬»Μ·―«μΩΘ§ΒΟΒΫ«≥ΜΤ…ΪΒΡ”ΆΉ¥ΈοΘ§άδ÷Ν “Έ¬ΚσΘ§Φ”»κΗ…‘οΒΡCH2Cl2 15 ml ΙΤδ»ήΫβΘ§Φ”»κ(3) 0.41 gΘ§≤ΔΒΈΦ”ΦΗΒΈΗ…‘οΒΡΏΝύΛΘ§‘Ύ “Έ¬œ¬ΫΝΑηΘ§»ή“Κ≥ ΉΊΚλ…ΪΓΘ6 hΚσΘ§TLCΦύ≤βΖ¥”ΠΈοΆξ»Ϊœϊ ßΓΘΦθ―Ι’τ»Ξ»ήΦΝΘ§”Ο≥Θ―Ι÷υ…ΪΤΉΖΫΖ®Ζ÷άκ≤ζΈοΘ§CHCl3Θ§ ·”ΆΟ―ΚΆMeOHΉςΧίΕ»œ¥Ά―ΓΘΒΟΒΫ≤ζΈο0.26 gΘ§≤ζ¬ ΈΣ42.1%Θ§Rf(D) 0.63Θ§‘ΣΥΊΖ÷ΈωC25H25N5O9(M618)Θ§ΦΤΥψ÷Β%ΘΚ C 58.04, H 4.91, N 13.22; ≤βΕ®÷Β%ΘΚ C 58.25Θ§ H 4.85Θ§ N 13.59ΓΘMS(FAB,M+1,540)ΓΘ1HNMR: ΠΡ 8.18(s,1H,2-H), 7.94(dd,2H,J=16 Hz,-CH=CH-), 7.49(m,5H,Harom), 6.12(1H,1ΓδH), 6.12(1H,2ΓδH), 5.77(m,1H,J=6.6 Hz,3ΓδH), 4.39(m,1H,J4Γδ3Γδ=6.6 Hz,4ΓδH), 4.49(dd,1H,Jab=12,Ja4Γδ=3.5 Hz,5Γδa), 4.28(dd,1H,Jb4Γδ=6.5 Hz,5Γδb), 2.12, 2.10, 2.06(s,9H,CH3CO)ΓΘ
, http://www.100md.com
4 6-»βΙπθΘΜυ-8-Εΰ»βΙπθΘΑΖΜυ-2Γδ,3Γδ,5Γδ-»ΐ““θΘΜυœΌή’(7)
(4) 0.31 g(0.76 mmol) 8-Α±Μυ-2Γδ,3Γδ,5Γδ-»ΐ““θΘΜυœΌή’[6]»ή”ΎΗ…‘οΒΡCH2Cl2»ή“Κ(10 ml)÷–Θ§ΒΈΦ”»κ»βΙπθΘ¬»0.51 g(3.04 mmol)ΒΡCH2Cl2»ή“Κ5 mlΘ§‘ΌΦ”»κΦΗΒΈ»ΐ““ΑΖΘ§‘Ύ “Έ¬œ¬ΦΧ–χΫΝΑη5 hΘ§¥ΥΜλΚœΈο”Ο≥Θ―Ι÷υ…ΪΤΉΖΫΖ®Ζ÷άκ(ΝΫ¥Έ)Θ§CHCl3Θ§ ·”ΆΟ―ΚΆMeOHΧίΕ»œ¥Ά―ΓΘΒΟΒΫ8Θ§6ΈΜ»ΐθΘΜ·≤ζΈο0.21 gΘ§≤ζ¬ ΈΣ34.7%Θ§Rf(D) 0.63Θ§‘ΣΥΊΖ÷ΈωC43H38N6O10Θ§ΦΤΥψ÷Β%ΘΚC 64.66Θ§H 4.76Θ§N 10.53;≤βΕ®÷Β%ΘΚC 65.09Θ§H 4.90Θ§N 10.26ΓΘMS(FAB,M+1,799), NMR: ΠΡ 8.71(s,1H,2-H), 7.98, 7.82, 7.75(dd,3H,JΠΝΠ¬=15.5 Hz,œ©ΜυΒΡHΠ¬), 6.77, 6.43(dd,3H,œ©ΜυHΠΝ), 6.56[s(br),1H,1ΓδH], 6.36[s(br),1H,2ΓδH], 6.13[s(br),1H,1ΓδH], 4.44(m,1H,J=3.36 Hz,5.92 Hz,4ΓδH), 4.47, 4.34(m,2H,5ΓδH), 2.18, 2.17, 2.06(s,9H,CH3CO)ΓΘ
, http://www.100md.com
5 6-(4-F-»βΙπθΘΜυ)-8-(4-F-»βΙπθΘΑΖΜυ)-2Γδ,3Γδ,5Γδ-»ΐ““θΘΜυœΌή’(8)
4-F-»βΙπΥα0.83 g(5 mmol)»ή”Ύ¬»Μ·―«μΩ10 ml÷–Θ§ΜΊΝς4 hΘ§Φθ―Ι’τ»ΞΕύ”ύΒΡ¬»Μ·―«μΩΘ§≤–”ύΈο”ΟΗ…‘οΒΡΏΝύΛ10 ml»ήΫβΘ§Φ”»κ8-Α±Μυ-2Γδ,3Γδ,5Γδ-»ΐ““θΘΜυœΌή’0.61 g(1.5 mmol)Θ§»ή“ΚΈΣΉΊΚλ…ΪΘ§ “Έ¬œ¬ΫΝΑη5 hΚσΘ§TLCΦύ≤βΖ¥”ΠΈο“―Ψ≠œϊ ßΘ§Φθ―Ι’τ»Ξ»ήΦΝΘ§ Θ”ύΈο”ΟEtOAc»ήΫβΘ§¬Υ»Ξ≤Μ»ήΈοΘ§¬Υ“Κ”Ο≥Θ―Ι÷υ…ΪΤΉΖ÷άκΘ§CHCl3Θ§ ·”ΆΟ―ΚΆMeOHΉςΧίΕ»œ¥Ά―ΓΘΒΟΒΫ8Θ§6ΈΜΕΰθΘΜ·≤ζΈο0.495 gΘ§≤ζ¬ ΈΣ46.3%Θ§Rf(D) 0.55ΓΘ‘ΣΥΊΖ÷ΈωC34H30F2N6O9.1/2H2OΘ§ΦΤΥψ÷Β%ΘΚ C 57.22Θ§ H 4.35Θ§ N 11.78; ≤βΕ®÷Β%ΘΚ C 57.20Θ§ H 4.57Θ§ N 11.52ΓΘMS(FAB,M+1,705), NMRΘΚΠΡ 8.89(s,1H,6-NH), 8.45(s,1H,2-H), 8.48,7.16(dd,2H,JΠ¬ΠΝ=15.4 Hz,Π¬-H), 6.62,6.52(dd,2H,ΠΝ-H), 7.53(8H,Harom), 7.04(8H,Harom), 7.04(1H,1ΓδH), 6.41(m,1H,2ΓδHΓδ), 6.14(brs,1H,3ΓδHΓδ), 4.44(1H,4ΓδHΓδ), 4.53(1H,5ΓδaH), 4.32(1H,5ΓδbH), 2.20(9H,CH3CO), 2.02(CH3CO)ΓΘ
, ΑΌΡ¥“Ϋ“©
6 6-(4-Cl-»βΙπθΘΜυ)-8-(4-Cl-»βΙπθΘΑΖΜυ)-2,Γδ3Γδ,5Γδ-»ΐ““θΘΜυœΌή’(9)
4-Cl-»βΙπΥα0.91 g(5 mmol)»ή”ΎΕ଻―«μΩ10 ml÷–,”Ύ90ΓφΜΊΝς5 hΚσΘ§Φθ―Ι’τ»Ξ Θ”ύΒΡΕ଻―«μΩΘ§ά以÷Ν “Έ¬Θ§”ΟΗ…‘οΒΡCH2Cl2 10 ml»ήΫβ≤–”ύΈοΘ§»ΜΚσΦ”»κ(4) 0.61 g(1.5 mmol)ΚΆ»ΐ““ΑΖ2 ml,‘Ύ “Έ¬œ¬ΫΝΑη5 hΓΘ»ή“ΚΈΣ…νΉΊΚλ…ΪΓΘΦθ―Ι’τ»Ξ»ήΦΝΘ§Φ”»κEtOAc»ήΫβΓΘΙΐ¬Υ≥ΐ»Ξ≤Μ»ήΈοΚσΘ§”Ο≥Θ―Ι÷υ…ΪΤΉΖ÷άκ(ΝΫ¥Έ)ΓΘ ·”ΆΟ―Θ§CHCl3ΧίΕ»œ¥Ά―ΓΘ”Ο““Υα““θΞΚΆ ·”ΆΟ―÷ΊΫαΨßΘ§ΒΟΒ≠ΜΤ…ΪΙΧΧε0.17 g,≤ζ¬ 15%ΓΘ Rf(D) 0.63ΓΘ‘ΣΥΊΖ÷ΈωC34H30Cl2N6O9.0.5H2O(M746), ΦΤΥψ÷Β%: C 54.69, H 4.16, N 11.26; ≤βΕ®÷Β%: C 54.70, H 4.23, N 11.48ΓΘ
, http://www.100md.com
7 6-±ΫΦΉθΘΜυ-8-±ΫΦΉθΘΑΖΜυ-2Γδ,3Γδ,5Γδ-»ΐ““θΘΜυœΌή’(10)
ΫΪΗ…‘οΒΡ8-Α±Μυ-2Γδ,3Γδ,5Γδ-»ΐ““θΘΜυœΌή’»ή”ΎΗ…‘οΒΡΏΝύΛ÷–Θ§ΒΈΦ”±ΫΦΉθΘ¬»0.21 ml,‘Ύ “Έ¬œ¬ΫΝΑη, 5 hΚσTLCΦύ≤βΖ¥”Π“―Άξ»ΪΓΘ ΫΪΜλΚœΈο’τΗ…Θ§”ΟΦθ―Ι÷υ…ΪΤΉΖ®Ζ÷άκΗςΉιΖ÷Θ§EtOAcΚΆΜΖΦΚΆιΉςΧίΕ»œ¥Ά―Θ§Rf(C) 0.72Θ§ΒΟ≤ζΈο0.27 gΘ§≤ζ¬ 60%ΓΘ‘ΣΥΊΖ÷Έω C30H27N6O9(M615)Θ§ΦΤΥψ÷Β%ΘΚ C 58.54, H 4.39, N 13.66; ≤βΕ®÷Β%ΘΚ C 58.73, H 4.57, N 13.94ΓΘ
8 6-Ε‘ΦΉ±ΫΦΉθΘΜυ-8-Ε‘ΦΉ±ΫΦΉθΘΑΖΜυ-2Γδ,3Γδ,5Γδ-»ΐ““θΘΜυ-œΌή’(11)
Ε‘ΦΉ±ΫΦΉΥα0.4 g(2.94 mmol)”ꬻ̷―«μΩ4 ml‘Ύ90ΓφΜΊΝς4 hΚσΘ§Φθ―Ι≥ΐ»ΞΙΐΝΩΒΡ¬»Μ·―«μΩΓΘΦ”»κΏΝύΛ5 ml»ήΫβ≤–ΝτΈοΓΘά以ΚσΘ§Φ”»κΒΫ8-Α±Μυ-2Γδ,3Γδ,5Γδ-»ΐ““θΘΜυœΌή’(4)0.408 g(1 mmol)ΒΡΈόΥ°ΏΝύΛ»ή“Κ(10 ml)÷–ΓΘ “Έ¬œ¬Ζ¥”Π4 hΓΘ≤ζΈο”Ο≥Θ―Ι÷υ…ΪΤΉΖ÷άκΘ§““Υα““θΞΚΆ ·”ΆΟ―ΧίΕ»œ¥Ά―ΓΘΒΟ≤ζΈο0.22 gΘ§≤ζ¬ 30.4%Θ§Rf(D) 0.57ΓΘ‘ΣΥΊΖ÷ΈωC32H32N6O9.C5H5NΘ§ΦΤΥψ÷Β%:C 61.41,H 5.12,N 13.55;≤βΕ®÷Β%:C 61.86,H 5.22,N 12.98ΓΘ1HNMR: ΠΡ 13.14(s,1H,8-NHCO), 9.04(s,1H,6-NHCO), 8.54(s,1H,2-H),7.97,7.35,7.26,8.26(m,10H,Harom), 6.62(1H,J1Γδ2Γδ=4.0 Hz,1ΓδH), 6.45(1H,J2Γδ1Γδ=4.0 Hz,J2Γδ3Γδ=6.5 Hz,2ΓδH), 5.96(1H,J3Γδ2Γδ=6.5 Hz,J3Γδ4Γδ=6.5 Hz,3ΓδH), 4.41(1H,J4Γδ3Γδ=6.5 Hz,3.5 Hz,5.5 Hz,4ΓδH), 4.51(m,1H,Jab=12.0 Hz,Ja4Γδ=3.5 Hz,5ΓδbH,5ΓδaH), 4.30(m,1H,Jba=12.0 Hz, Jb4Γδ=5.5 Hz)ΓΘ
, ΑΌΡ¥“Ϋ“©
9 6-±ΫΦΉθΘΜυ-8-±ΫΦΉθΘΑΖΜυœΌή’(12)
(10) 0.225 g(0.366 mmol)»ή”ΎCH3ONa/MeOH 4 ml(0.1 mol.L-1)»ή“ΚΚΆΗ…‘οMeOH 10 ml÷–, ‘Ύ “Έ¬œ¬Ζ¥”Π10 hΓΘ”ΟœΓ¥ΉΥα÷–ΚΆ¥ΥΜλΚœΈοΘ§»ΜΚσ”ΟΦθ―Ι÷υ…ΪΤΉΖ®Ζ÷άκΘ§ΒΟΒΫ(12) 60 mgΘ§≤ζ¬ ΈΣ33.5%ΓΘRf(D) 0.41ΓΘ‘ΣΥΊΖ÷ΈωC24H21N6O6(M489), ΦΤΥψ÷Β%: C 58.90, H 4.29, N 17.18; ≤βΕ®÷Β%: C 58.59, H 4.17, N 16.93ΓΘ1HNMR: 12.75(s,1H,8-NH), 11.75(s,1H,6-NH), 8.71(s,1H,2-H), 8.23,7.56(m,10H,Harom), 6.40(d,1H,J1Γδ2Γδ=5.5 Hz,1ΓδH), 5.20(T,1H,J2Γδ1Γδ=5.5 Hz,2ΓδH), 4.38(t,1H,J2Γδ3Γδ=5.5 Hz,J3Γδ4Γδ=9 Hz,3ΓδH), 3.98(m,1H,J4Γδ5Γδ=5.5 Hz,4ΓδH), 3.73(m,1H,Jab=12 Hz,Ja4Γδ=4.5 Hz,5ΓδaH), 3.56(m,1H,JbΓδ4Γδ=5.5 Hz,5ΓδbH)ΓΘ
, ΑΌΡ¥“Ϋ“©
10 2Γδ,3Γδ-‘≠ΦΉΥα““θΞΜυ-8-Cl-œΌή’(13)
8-Cl-œΌή’2 g(6.62 mmol)ΚΆ‘≠ΦΉΥα““θΞ5.3 mlΘΜ»ΐ¬»““Υα4.66 gΚΆΕΰ―θΝυΜΖ53 mlΒΡΜλΚœΈο‘Ύ60ΓφΦ”»»Ζ¥”Π4 hΘ§TLCΦύ≤βΖ¥”ΠΆξ»Ϊ[7]ΓΘ”ΟCH2Cl2 100 mlœΓ Ά¥Υ»ή“ΚΘ§»ΜΚσ”ΟNaCO3»ή“ΚΚΆH2OΗςœ¥3¥ΈΓΘΗ…‘οΘ§≥ΐ»Ξ»ήΦΝΚσΘ§”ΟΈόΥ°EtOH÷ΊΫαΨßΘ§ΒΟΒΫ≤ζΈο2.11 gΘ§≤ζ¬ 89.2%ΓΘNMR÷Λ ΒΈΣΖ«Ε‘”≥“λΙΙΧεΜλΚœΈοΘ§ Rf(D) 0.87ΓΘ1HNMR: “λΙΙΧεI,ΠΡ 8.19(s,1H,2-H), 7.57(s,2H,6-NH2), 6.23(s,1H,‘≠ΦΉΥα““θΞΒΡH), 6.20(d,1H,J1Γδ2Γδ=2.7 Hz,1ΓδH), 5.78(dd,1H,J1Γδ2Γδ=2.7 Hz,J2Γδ3Γδ=6.6 Hz,2ΓδH), 5.14(m,1H,J3Γδ4Γδ=3.6 Hz,3ΓδH), 4.28(m,1H,J4Γδ5Γδ=6.6 Hz,4ΓδH), 3.50ΓΪ3.37(m,2H,5ΓδH), 4.21(m,2H,J=6.6 Hz,-CH2-), 1.22(t,3H,-CH3), 5.16ΓΪ5.01(br,1H,5Γδ-OH)ΓΘ“λΙΙΧεIIΘ§ ΠΡ 8.17(s,1H,2-H), 7.57(s,2H,6-NH2), 6.17(s,1H,‘≠ΦΉΥαθΞΒΡH), 6.07(s,1H,1ΓδH), 5.73(dd,1H,J1Γδ2Γδ=3 Hz,2ΓδH), 5.02(m,1H,J3Γδ4Γδ=3.6 Hz,3ΓδH), 4.19(m,1H,J4Γδ5Γδ=3.8 Hz,4ΓδH), 3.50ΓΪ3.37(m,1H,5ΓδH), 4.21(m,2H,J=6.6,-CH2-), 1.13(t,3H,CH3), 5.16ΓΪ5.01(m,1H,5Γδ-OH)ΓΘ
, ΑΌΡ¥“Ϋ“©
11 2Γδ,3Γδ-±ϊ≤φΜυ-8-Cl-œΌή’(14)
8-Cl-œΌή’0.3 g(1 mmol)”ΎΤ’Ά®±ϊΆΣ÷–ΨγΝ“ΫΝΑηΘ§‘Ύ±υ‘Γά以œ¬Θ§Ζ÷≈ζΒΈΦ”ΝρΥα0.15 mlΓΘΈ§≥÷Ζ¥”ΠΈ¬Ε»‘Ύ(0.5ΓΪ10)ΓφΓΘΒΈΦ”Άξ±œΚσΘ§‘ΌΦΧ–χΫΝΑη5 hΓΘ‘Ύ¥ΥΤΎΦδΘ§Έ¬Ε»÷πΫΞ…ΐ÷Ν “Έ¬ΓΘΖ¥”ΠΆξ≥…ΚσΘ§‘Ύ±υ‘Γά以œ¬Θ§”ΟNaOH»ή“Κ÷–ΚΆ÷Ν÷––‘Θ§Ιΐ“ΙΘ§Ιΐ¬Υ≥ΐ»Ξ―ΈΘ§¬Υ“Κ≈®ΥθΚσ”Ο¬»Ζ¬»ήΫβΘ§Υ°œ¥¥Υ¬»Ζ¬»ή“ΚΘ§≤Δ”Ο≥Θ―Ι÷υ…ΪΤΉΖ®Ζ÷άκ≥ω≤ζΈο0.32 g, Rf(C) 0.89,‘ΣΥΊΖ÷ΈωC13H16ClN5O4(M342)Θ§ΦΤΥψ÷Β%: C 45.61, H 4.68, N 20.47; ≤βΕ®÷Β%: C 45.24, H 4.47, N 20.24ΓΘ
12 2Γδ,3Γδ-±ϊ≤φΜυ-5Γδ-O-ΦΉΜ«θΘΜυ-8-Cl-œΌή’(15)[8]
, ΑΌΡ¥“Ϋ“©
ΫΪ(14) 1 g(2.93 mmol)»ή”ΎΗ…‘οΏΝύΛ30 ml÷–Θ§‘Ύ±υ‘Γά以œ¬Θ§ΒΈΦ”ΦΉΜ«θΘ¬»0.5 mlΓΘ»ή“ΚΦ¥±δΜΤ…ΪΓΘ‘Ύ¥ΥΈ¬Ε»œ¬ΫΝΑη4 hΓΘΦθ―Ι’τ»Ξ»ήΦΝΘ§”ΟΦθ―Ι÷υ…ΪΤΉΖ®Ζ÷άκΓΘEtOAcΚΆEtOHΧίΕ»œ¥Ά―Θ§ΒΟ≤ζΈο0.39 gΘ§≤ζ¬ 31.7%Θ§Rf(B) 0.78ΓΘ
13 5Γδ-O-ΦΉΜ«θΘΜυ-8-Cl-œΌή’(16)
ΫΪ(15) 0.21 g(0.5 mmol)»ή”ΎHCOOH(80%) 2.5 ml÷–Θ§ “Έ¬œ¬ΫΝΑη10 hΓΘ»ΜΚσΦθ―Ι’τ»ΞHCOOHΘ§≤Δ”ΟΈόΥ°EtOH÷Ί’τ3¥ΈΓΘ≤–”ύΈο”Ο≥Θ―Ι÷υ…ΪΤΉΖ®Ζ÷άκΓΘ ·”ΆΟ―ΚΆ““Υα““θΞΉςΧίΕ»œ¥Ά―Θ§ΒΟΒΫΑΉ…ΪΙΧΧε0.13 g,≤ζ¬ 68%Θ§‘ΣΥΊΖ÷ΈωC11H14ClN5O6S(M380),ΦΤΥψ÷Β%:C 34.74,H 3.68,N 18.42;≤βΕ®÷Β%:C 34.97,H 3.72,N 18.19ΓΘ
, ΑΌΡ¥“Ϋ“©
14 2Γδ,3Γδ-±ϊ≤φΜυ-5Γδ-O-œθΜυ-8-Cl-œΌή’(17)
ΫΪ≈®HNO3 1.4 mlΒΈΦ”»κ±υ―Έ‘Γά以ΒΡΒ»ΧεΜΐΒΡ““Υατϊ÷–Θ§ΒΟΒΫΜλΚœΥατϊΓΘ(14) 0.26 g(0.7 mmol) »ή”ΎEtOAc÷–Θ§”Ο±υ―Έ‘Γά以÷Ν-30ΓφΓΘ»ΓΜλΚœΥατϊ1 mlΒΈΦ”»κ(14)ΒΡ»ή“Κ÷–Θ§Ζ¥”ΠΜλΚœΈο‘Ύ¥ΥΈ¬Ε»œ¬ΦΧ–χΖ¥”ΠΘ§4.5 hΚσΘ§”ΟNaHCO3»ή“Κ÷–ΚΆΓΘ”–Μζ≤ψΚΆH2O≤ψΖ÷ΩΣΘ§H2O≤ψ”ΟEtOAcίΆ»Γ3¥ΈΓΘΚœ≤ΔEtOAc»ή“ΚΘ§Η…‘οΘ§”ΟΦθ―Ι÷υ…ΪΤΉΖ®ΒΟΒΫ0.2 g≤ζΈο(ΑΉ…ΪΙΧΧε)Θ§≤ζ¬ 74.1%,Rf(C) 0.44ΓΘ‘ΣΥΊΖ÷ΈωC13H15ClN6O6.H2O(M387),ΦΤΥψ÷Β%ΘΚ C 40.31, H 3.88, N 21.71ΘΜ ≤βΕ®÷Β%ΘΚ C 39.84, H 3.96, N 21.46ΓΘ
, ΑΌΡ¥“Ϋ“©
15 (17)ΒΡ»Ξ±ΘΜΛΖ¥”Π5Γδ-O-œθΜυ-8-Cl-œΌή’(18)
ΫΪ(17)»ή”Ύ88%ΒΡHCOOH÷–Θ§‘Ύ “Έ¬œ¬ΫΝΑη10 hΘ§Φθ―Ι’τ»ΞHCOOHΘ§‘Ό”ΟΈόΥ°EtOH÷Ί’τ3¥ΈΓΘΖ¥”ΠΜλΚœΈο”Ο≥Θ―Ι÷υ…ΪΤΉΖ®Ζ÷άκΓΘEtOAcΚΆ ·”ΆΟ―ΧίΕ»œ¥Ά―ΓΘΒΟΒΫ≤ζΈο60 mgΘ§≤ζ¬ 61.8%ΓΘRf(B) 0.74Θ§‘ΣΥΊΖ÷ΈωC10H11ClN6O6(M347), ΦΤΥψ÷Β%ΘΚ C 34.58, H 3.17, N 24.21ΘΜ ≤βΕ®÷Β%ΘΚ C 34.83, H 3.32, N 23.86ΓΘ
16 5Γδ-¬»-5Γδ-»Ξ―θ-2Γδ,3Γδ-―«μΩΜυ-8-Cl-œΌή’(19)
ΫΪ(13)¥ζΧφ8-Cl-œΌή’”κHMPAΚΆSOCl2ΒΡΜλΚœΈοΖ¥”ΠΓΘΖ¥”ΠΜλΚœΈο”ΟΦθ―Ι÷υ…ΪΤΉΖ÷άκΘ§““Υα““θΞΓΔΜΖΦΚΆιΚΆEtOHΧίΕ»œ¥Ά―,ΒΟΒΫ≤ζΈο0.2 gΘ§≤ζ¬ 32.8%ΓΘRf(F) 0.78ΓΘ‘ΣΥΊΖ÷ΈωC10H9Cl2N5O4S.H2O(M370.5), ΦΤΥψ÷Β%ΘΚ C 32.38, H 2.56, N 18.89ΘΜ ≤βΕ®÷Β%ΘΚ C 32.78, H 2.53, N 18.57ΓΘ1HNMRΘΚ ΠΡ 6.43(d,1H,J1Γδ2Γδ=2.4 Hz,1ΓδH), 6.39(m,1H,J1Γδ2Γδ=2.4 Hz,J2Γδ,3Γδ=7.5 Hz,2ΓδH), 5.87(m,1H,J2Γδ,3Γδ=7.5 Hz,J3Γδ,4Γδ=3.9 Hz,3ΓδH), 4.73(m,1H,J=3.9,6.0,7.2 Hz,4ΓδH), 3.86(m,2H,Jab=11.1, Ja4Γδ=7.2 Hz,Jb4Γδ=6.3 Hz,5ΓδH), 7.64(s,2H,6-NH2), 8.19(s,1H,2-H)ΓΘ
, ΑΌΡ¥“Ϋ“©
17 5Γδ-¬»-5Γδ-»Ξ―θ-8-Cl-œΌή’(20)[9]
ΝυΦΉΜυΝΉθΘΑΖ(HMPA)1.6 ml”Ο±υ‘Γά以ȧœρΤδ÷–ΒΈΦ”SOCl2 0.24 mlΓΘ‘Ύ0ΓφΫΝΑηœ¬15 minΚσΘ§ΆΕ»κ8-Cl-œΌή’0.3 g(1 mmol)ΓΘΖ¥”Π2 hΓΘ “Έ¬œ¬ΫΝΑηΙΐ“ΙΓΘ»ή“Κ≥ œ Κλ…ΪΓΘΫΪΜλΚœΈο«ψ»κ±υH2O 25 ml÷–Θ§NaHCO3»ή“Κ÷–ΚΆ÷Ν÷––‘Θ§”ΟΦθ―Ι÷υ…ΪΤΉΖ÷άκΘ§EtOAcΚΆEtOHΧίΕ»œ¥Ά―ΓΘΒΟΒΫΑΉ…ΪΙΧΧε0.1 gΘ§≤ζ¬ 31.3%,Rf(B) 0.90ΓΘ‘ΣΥΊΖ÷ΈωC10H11Cl2N5O3Θ§ΦΤΥψ÷Β%ΘΚ C 37.50, H 3.44, N 21.88ΘΜ ≤βΕ®÷Β%ΘΚ C 37.87, H 3.78, N 21.48ΓΘ
÷¬–Μ Άθ ι”ώΫΧ ΎΧαΙ©»Γ¥ζ»βΙπΥαΘΜ÷ΊΒψ Β―ι “ΒΡ‘ΣΥΊΖ÷Έω Β―ι “ΚΆΚΥ¥≈ “Θ§÷––Ρ Β―ι “÷ ΤΉ “Θ§±±Ψ©¥σ―ßΚΆΖάΜ·‘ΚΚΥ¥≈ “Θ§÷–ΙζΩΤ―ß‘ΚΜ·―ßΥυ–≠ΉςΆξ≥…ΝΥΚΥ¥≈Ι≤’ώΘ§÷ ΤΉΚΆ‘ΣΥΊΖ÷ΈωΘΜ÷ΊΒψ Β―ι “Άθ»π«δΒ»άœ ΠΆξ≥…≥θ≤Ϋ…ζΈοΜν–‘…Η―Γ Β―ιΓΘ
, ΑΌΡ¥“Ϋ“©
*ΝΣœΒ»Υœ÷÷ΖΘΚΨϋ ¬“Ϋ―ßΩΤ―ß‘ΚΕΨΈο“©Έο―–ΨΩΥυΘ§±±Ψ© 100850
Tel:(010)66887429-66611
≤ΈΩΦΈΡœΉ
1 ΖΫΦ“¥ΜΘ§ ·”άΫχΘ§≈μΟτΘ§Β». 8-¬»œΌή’ΒΡΩΙ÷ΉΝωΉς”Ο―–ΨΩ. ÷–ΜΣ÷ΉΝω‘”÷Ψ, 1995,17ΓΟ5
2 ΆθœήλβΘ§÷ΘΒ¬œ»Θ§ΝθνΘΘ§Β». 8-¬»œΌή’”’ΒΦMOLT-4œΗΑϊΒρΆω. ΩΤ―ßΆ®±®, 1997,42ΓΟ189
3 Lin Z, Nadzan AM, Heyman RA, et al. Discovery of novel retinoicacid receptor agonists having potent antiproliferative activity in cervical cancer cells. J Med Chem, 1996,39ΓΟ2659
, ΑΌΡ¥“Ϋ“©
4 Holy A, Rosenberg I. An improved synthesis of S-adenosyl-L-nomocystein and related compounds. Collect Czech Chem Commun, 1985,50ΓΟ1514
5 Long RA, Robins RK, Townsend LB. Purine nucleosides, XV. The synthesis of 8-amino- and 8-substituted aminopurine nucleosides. Biochemistry, 1967,32ΓΟ2751
6 Ikehara M, Yamada S. Studies of nucleosides and nucleotides. XLIX. Synthesis of 8-fluoroadenosine. Chem Pharm Bull, 1971,19ΓΟ104
, http://www.100md.com
7 Eckstein F, Cramer F. Notize uber 2Γδ,3Γδ-O-athoxy-methylen-derivate von Ribonucleosiden. Chem Ber, 1965ΓΟ995
8 Sharma M, Wikiel H, Hromchak R, et al. Synthesis of 5Γδ-substituted derivatives of the pyrrolo[2,3-d]-pyrimidine nucleoside sangivamycin and their effect on protein kinase A and C. Nucleosides and Nudeotides, 1993,12(3 & 4)ΓΟ295
9 Prisbe EJ, Smejkal J, Verheyden JPH, et al. Halo sugar nucleosides. 5. Synthesis of angustmycin A and some base analogues. J Org Chem, 1976,41ΓΟ1836
’Ηε»’ΤΎ: 1997-11-28, ΑΌΡ¥“Ϋ“©(ΚΈΨϋΝ÷* ’≈άώΚΆ)