邵峰:从天然免疫到癌症,细胞焦亡扮演何种角色?
文/《中国医药科学》记者 费 菲
2015年12月7日,43岁的独立研究者邵峰当选中国科学院生命科学和医学学部院士,刷新了我国两院院士年龄榜,是1600多名院士中最年轻的一位。
1996年邵峰毕业于北京大学技术物理系应用化学专业,1999年中科院生物物理所硕士毕业,2003年获得美国密歇根大学医学院博士学位,2005年在哈佛大学医学院完成博士后训练后回国,在北京生命科学研究所创建实验室,带领团队开展独立研究生涯,在“细胞焦亡”领域做出了许多重要的开创性工作。回国前,邵峰在世界顶级学术期刊《细胞》(cell)和《科学》(science)杂志上分别发表了1篇论文。2005年回国后至今以通讯作者身份在《自然》(nature)、science、cell共发表论文11篇,研究成果多次获权威专家和学术杂志重点评述。目前邵峰实验室的研究继续保持着高水准,每一个成果都吸引了全球大量研究者的目光。
2013年,邵峰院士作为首位大陆本土科学家荣获国际蛋白质学会颁发的鄂文・西格青年科学家奖。2015年,他成为欧洲分子生物学组织(EMBO)外籍成员,此前只有5位中国科学家获选。他还曾先后荣获霍华德・休斯研究所青年科学家奖、吴阶平―保罗・杨森医学药学奖、周光召杰出青年基础科学奖和国家杰出青年基金。截至目前,邵峰实验室已发表学术论文70多篇,同行引用6000多次。
, http://www.100md.com
2017年,担任北京生命科学研究所(NIBS)学术副所长、资深研究员的邵峰院士带领团队首次揭示和阐明了细胞焦亡的机制。他们的研究结果显示,在细胞焦亡的发生机制中,半胱天冬酶(caspase)炎症小体下游的Gasdermin家族蛋白可能发挥了关键作用。
在2017年中国免疫学会年会上,邵峰院士深入浅出地阐述了细胞焦亡对人体免疫系统和癌症发病的相关机制。
他们在体外试验将小鼠骨髓诱导的巨噬细胞感染沙门氏杆菌(胞内菌)或痢疾杆菌(胞内自由生存的革兰氏阴性菌),同时将白细胞介素-1β(IL-1β)和染料加入培养基。感染后染料进入细胞核,免疫荧光显微镜观察到整个细胞发生爆炸性、裂解性死亡,这就是细胞焦亡(pyroptosis)。这一现象早在1992年就被观察到了,Nature杂志刊登了Zychlinsky A等的文章《弗氏志贺氏杆菌诱导感染的巨噬细胞凋亡》(Shigella flexneri induces apoptosis in infected macrophages),指出弗氏志贺氏杆菌可以诱导感染的宿主巨噬细胞发生程序性死亡。当时对程序性细胞死亡方式的认识仅限于凋亡,因此将其归结为细胞凋亡作用,这种认识是错误的。几年后的研究发现这种细胞死亡过程需要依赖半胱天冬酶-1(caspase-1)且伴随炎症发生,导致在2000年前后大家误将细胞凋亡和半胱氨酸蛋白酶 (caspase) 家族联系起来。但显然这一结论并不正确。这一细胞持续性死亡会发生细胞膜破裂,而细胞凋亡的细胞膜是不破裂的。 在20世纪90年代,人们对这种新的细胞死亡方式的性质不甚理解,并在较长时间内与细胞凋亡相混淆。
, 百拇医药
□邵峰教授在实验室
炎性caspase-1是怎么激活的?2002年瑞士Tschopp研究小组首次提出了炎症小体(Inflammasome)的概念,虽然没有提出实验证据。邵峰实验室的研究认为,炎症小体复合物能够激活胱冬肽酶-1(caspase-1),促进炎性细胞因子前体白介素-1(IL-1Lβ)和白介素-18(IL-18)切割成熟,同时激活的Caspase-1还可以使细胞发生焦亡。炎症小体在过去十几年里成为天然免疫领域最受关注的热点问题。各类炎症小体的区别在于不同的NLR 蛋白可以感受细胞内病原或危险信号的感染。其实当时没有实验证据存在炎症小体的复合物,更多是一种概念的提法,但确实有些疾病的证据支持这一说法,因为很多NLR家族基因的突变会导致一些自身炎症性疾病(AIDs)或自身免疫病。自身炎症性疾病主要是天然免疫系统发生问题,导致白介素-1(IL-1)活化。自身免疫病则是刺激人体免疫系统产生攻击自身的抗体。
邵峰院士介绍,我们实验室最早时探究NLR蛋白是怎么识别和清除细胞内的病原细菌。天然免疫反应的第一步是通过感知蛋白(受体蛋白)最先感知到细菌的入侵,此前对受体蛋白的研究主要集中在细胞膜上的一类天然免疫受体,因此,我们致力于发现宿主细胞质中感知细菌和病原细菌的感知蛋白(受体蛋白)的分子免疫机制,细菌内毒素(即脂多糖,LPS)被识别及其诱导炎症性坏死的机制是什么?我们创建了一种能将细菌内毒素(即脂多糖,LPS)高效导入哺乳动物细胞内的转染方法,发现LPS的胞内天然免疫受体是炎症性半胱天冬酶(caspase),并找到了全新的识别细菌主要模式分子的天然免疫受体(感知蛋白)NAIPs、Pyrin。其中,NAIPs蛋白可作为炎症小体的受体,识别细胞内细菌的鞭毛素蛋白和内毒素分泌系统的结构成分。Pyrin蛋白的主要功能是间接感知各种细菌毒力的作用,胞内的Rho家族小G蛋白修饰和失活导致肌动蛋白细胞骨架发生紊乱,进而活化经典炎症小体通路中的caspase-1。
, http://www.100md.com
20世纪90年代,Bruce Beutler等鉴定出Toll样受体4(TLR4)为细胞外的LPS的膜上受体,LPS结合TLR4后诱导细胞因子和炎症因子转录。邵峰由此产生了一个疑问:在细胞质里是否有受体蛋白来识别病原菌最为重要的模式分子LPS?两年前,邵峰实验室发现了一个炎症性Caspase家族,人细胞中的Caspase-4、Caspase-5是小鼠非经典炎症小体通路中Caspase-11的同源蛋白。与caspase-1类似的caspase-11可直接作为小鼠胞内受体识别革兰氏阴性菌的脂多糖(LPS),同时可诱导细胞焦亡的发生。
Toll样受体家族(NLRs)基因的突变还会导致自身炎症性疾病或自身免疫性疾病,这里面存在因果关系。比如NLR3基因突变可导致痛风、NLRP3突变可导致家族性寒冷性自身炎症综合征(FCAS)和穆一韦二氏综合征(MWS)、慢性婴儿神经皮肤关节综合征(CINCA)/NOMID(新生儿多系统炎症性疾病)、NLRC4激活的炎症小体突变可导致小肠结肠炎综合征和自身炎症(syndrome of enterocolitis and autoinflammation),也可导致自身免疫性疾病――复发性巨噬细胞活化综合征(recurrent macrophage activation syndrome)。MEFV基因编码的Pyrin功能获得性突变可导致自身炎症性疾病――家族性地中海热(FMF)。感知蛋白NAIPs、Pyrin的正常功能是什么?就是识别细菌的结构成分――脂多糖、鞭毛素蛋白。基因突变就是在未感染细菌时已被激活,从而导致各种疾病。这些炎症小体的基因突变会导致自身炎症性疾病,在自身免疫病发生中也起到辅助性作用。
, 百拇医药
细胞焦亡的“促成者”――GSDMD蛋白
细胞焦亡(pyroptosis)与细胞凋亡(apoptosis)究竟有什么差别?小鼠的巨噬细胞发生细胞凋亡时,细胞膜骤缩内陷裂成由细胞膜包裹的一个个泡状小体(即凋亡小体),凋亡小体表面有特殊信号导致其很快被巨噬细胞或邻近细胞识别并吞噬,不释放细胞内容物,因此不会导致组织水平的损伤和炎症反应,细胞的这种干净死亡(clean death)方式不影响其他细胞的正常功能。细胞焦亡是细胞体积不断胀大至细胞膜破裂,释放细胞内容物从而激活强烈的炎症反应,属于一种细胞炎性坏死(dirty death方式)。caspase-1/11都是在天然免疫的下游感知病原细菌的感染。caspase-1/11导致细胞焦亡发生的机制是什么?2015年,邵峰研究组运用全基因组编辑技术筛选人类基因组中的近2.2万个基因后,成功识别出一个新的基因――导致细胞焦亡的关键蛋白GSDMD并阐述了相关机制。GSDMD是所有炎性caspase的共有底物,其编码的蛋白质大约有500多个氨基酸和2个结构域氨基端gasdermin-N和羧基端gasdermin-C。正常状态下两个结构域相互“勾连”作用,处于无活性的自抑制状态,当炎性caspase-1或caspase-11活化后,可特异性切割GSDMD蛋白2个结构域之间的连接区域,释放的N端结构域可识别并结合细胞膜上的磷脂类分子,在细胞膜上打孔,导致细胞渗透压变化,细胞膜胀大裂解引发细胞焦亡。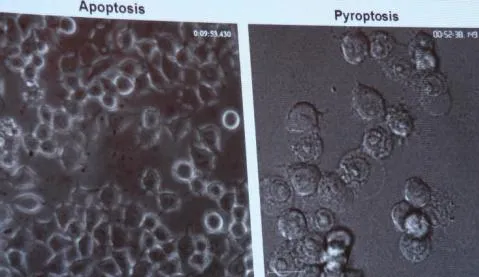
, 百拇医药
细胞凋亡(左图)和细胞焦亡(右图)
由此可知,GSDMD蛋白与细胞焦亡密切相关,GSDMD蛋白是caspase-1和caspase-11/4/5诱导细胞焦亡的执行者,也清楚地阐述了细胞焦亡的本质是细胞炎性坏死。如果在细胞中去除GSDMD蛋白,细胞便不再发生细胞焦亡。长期以来人们都不甚清楚白细胞介素-1β(IL-1β)释放至细胞外的机制,邵峰团队的研究指出,GSDMD蛋白的缺失不会抑制激活caspase-1和对下游白介素1β的切割。在敲除GSDMD的细胞中,白细胞介素-1β可以被caspase-1切割成熟,但切割后成熟的白介素1β却几乎不能分泌到细胞外,显示细胞焦亡对于白介素1β的分泌必不可少,IL-1β不是通过分泌信号释放到细胞外,而是GSDMD在细胞膜上打孔后排出,GSDMD可能是IL-1β排出细胞外的必要条件。敲除GSDMD基因可以阻止白介素-1β的分泌和炎症小体及细菌LPS引起的细胞焦亡,使细胞转而发生凋亡。
caspase参与的炎症反应及细胞焦亡能有效提高机体抵抗内源性和外源性刺激的能力, 达到保护宿主的目的。caspase-1 和caspase-4/5/11介导的细胞焦亡通路具有天然免疫功能,当宿主细胞发生感染时可识别细菌鞭毛素蛋白、LPS,激活炎症小体介导的抗细菌天然免疫反应,当病原被天然免疫系统识别后启动焦亡。因此,将天然免疫受体基因敲除的小鼠无法清除感染的细菌而死亡,而正常野生型小鼠可以清除革兰氏阴性菌的感染而得以存活。实验中观察到,敲除GSDMD基因的小鼠脾脏和肝脏中,可以发现大量细菌的复制和病变。
, http://www.100md.com
另一方面,天然免疫系统也是双刃剑,细胞焦亡通路的过度活化也会引起负面反应。邵峰教授指出,革兰氏阴性菌导致的脓毒症是过度的细胞焦亡导致的。在小鼠模型注射大量LPS,它会在一天内死亡;如果把小鼠胞内受体caspase-11或下游的GSDMD敲除则可以存活。实验室还制作了一个抗体,只识别切割后的GSDMD蛋白,现在需要临床样本来验证这一抗体,有兴趣的临床医生可以与实验室取得联系。GSDMD蛋白是被caspase一分为二,一旦切割成功,细胞一定会发生凋亡。
邵峰实验室进一步研究发现,GSDMD代表了一个被称为gasdermin的家族性蛋白,除GSDMD外,GSDMA、GSDMB、GSDMC、GSDME(此 前 称DFNA5)、DFNB59等都具有N端和C端2个结构域,并能被炎性caspase切割。Gasdermin的家族蛋白中,仅GSDMD的2个结构域中间有caspase-1和-11的切割位点,但所有gasdermin家族性蛋白N端结构域被活化后都可在细胞膜上打孔从而诱导细胞焦亡。和GSDMD类似,未感染时这些蛋白N端和C端通过自抑制作用保持无活性状态。正因为此,邵峰团队最近将细胞焦亡重新定义为 “由Gasdermin家族蛋白介导的程序性细胞坏死”。由于GSDMD与炎症小体信号通路重要分子caspase-1和caspase-11/4/5相关,但其他Gasdermin家族蛋白与其无关,需要通过其他机制被切割活化,但最终也会通过启动细胞焦亡激活天然免疫反应。
, 百拇医药
邵峰实验室又在细胞焦亡研究领域取得了新的突破,揭示了另一种Gasdermin家族蛋白GSDME引起细胞焦亡的机制,研究结果已在线发表在2017年5月1日Nature杂志上。一组将实验用增殖表皮癌细胞(Hela Cells)中敲除GSDMD蛋白,回补GSDMD+肿瘤坏死因子a(TNFa),细胞遂发生了凋亡。另一组将Hela细胞的GSDMD敲除,回补一个改造过的可以被caspase-3切割活化的GSDMD(在两个结构域中间的caspase-1/4/11切割位点突变为caspase-3的切割位点,由于肿瘤坏死因子a诱导细胞凋亡是通过活化caspase-3),细胞从凋亡转化为焦亡,说明GSDMD蛋白只要在两个结构域中切割就会诱导细胞凋亡,这一发现对癌症治疗(尤其是化疗)的研发具有重要的指导意义。实验也说明细胞焦亡比细胞凋亡发生得更快。在第一组实验中可以观察到caspase-3切割活化的GSDMD,但另一组实验却观察不到caspase-3活化的GSDMD形态,这是由于焦亡发展得太快,掩盖了这一激活的过程,即细胞来不及凋亡就发生了焦亡。
, http://www.100md.com
邵峰实验室还完成了Gasdermin家族所有蛋白的对照实验。此前将GSDMD的caspase-1/4/11切割位点突变为caspase-3切割位点,可以中止TNFa诱导活化caspase-3引起的HeLa细胞凋亡,并转换为焦亡。在对照实验中,HeLa细胞中表达野生型的GSDME也同样可以将凋亡转换为焦亡,这是为什么呢?通过分析蛋白序列后发现,GSDME的N端和C端结构域中间存在天然的caspase-3切割位点。将caspase-3切割位点D突变为A,可以抑制caspase-3切割N端和C端结构域,细胞则恢复凋亡信号,不再发生焦亡。通过蛋白印迹实验(Westren Blot)中也证实,如果把caspase-3敲除或把切割位点D突变,就不能再切割N端和C端结构域,细胞焦亡也就不会发生。
另外一个有趣的现象是,肿瘤坏死因子a诱导细胞凋亡是通过活化caspase-3实现的,而caspase-3可以通过多种机制被活化。最经典的caspase-3活化机制是DNA损伤通过线粒体释放凋亡诱导因子,参与激活caspase-3。化疗药物顺铂等都会通过损伤DNA导致caspase-3活化。和GSDMD类似,caspase-3切割释放的GSDME的N端片段,能特异性结合4,5-二磷酸磷脂酰肌醇[PI(4,5)P2]并导致含有该磷脂的脂质体泄漏,负染电镜结果显示切割活化的GSDME可以在脂质体膜上打孔。这些结果说明GSDME是由caspase-3切割后活化,进而在膜上打孔并触发细胞焦亡。教科书上说caspase-3细胞凋亡,本质上是细胞炎性坏死。绝大多数常用的(癌)细胞系均不表达GSDME蛋白,所以被观察到的细胞确实是发生了凋亡。
, 百拇医药
邵峰实验室只找到了两株癌细胞表达高水平的内源GSDME――人的神经母细胞瘤细胞SH-SY5Y和恶性黑色素瘤细胞MeWo。把GSDME表达和不表达的细胞用化疗药物处理后,不表达GSDME的细胞用各种损伤DNA的化疗药物诱导后均发生了凋亡,而所有高表达GSDME的细胞都发生了焦亡。进一步检测了NCI-60(在培养基中生长的标准60种人类癌细胞系)的全部60株癌细胞后,邵峰实验室研究者发现,大多数的细胞不表达或低表达GSDME,仅有4~5个细胞表达较高水平的GSDME。查找现有文献发现,GSDME基因的启动子在癌化过程会发生DNA甲基化,导致表观遗传沉默。邵峰实验室于是做了一个很有意思的实验。不表达GSDME的癌细胞用地西他滨(目前唯一获准上市的DNA甲基化转移酶抑制剂地西他滨,可阻断DNA甲基化过程)处理后,成功诱导GSDME表达,此时再用传统化疗药物治疗,癌细胞对传统化疗药物变得更敏感(焦亡关键因子GSDME表达),更易发生炎性坏死。
很多临床医师开展的实验证明,地西他滨与传统化疗药物结合用于治疗骨髓增生异常综合征(MDS)效果更好,但找不到其中的机制。邵峰实验室的这项研究结果恰好解释了这一机制。小鼠的很多正常组织器官高表达GSDME,而癌化的细胞由于DNA甲基化介导的表观遗传沉默对焦亡关键因子GSDME不表达,使用地西他滨后激活caspase-3后诱导GSDME表达,从而促使癌细胞发生GSDME依赖的焦亡。
, 百拇医药
邵峰实验室进一步检测了5株人体正常组织来源的原代细胞血管内皮细胞,除2株不表达GSDME,另外3株高表达GSDME。经化疗药物处理这3株原代细胞后,GSDME高表达的细胞伴随caspase-3依赖的GSDME切割,发生焦亡;不表达GSDME的2株原代细胞则发生凋亡。在GSDME高表达的原代细胞中,用RNA干扰(RNAi)敲低GSDME表达后,化疗药物引起的细胞焦亡则转换为凋亡。
癌症患者使用化疗药物后为何会产生很大的毒副作用?因为在使用化疗药物后,正常原代细胞可高表达焦亡因子GSDME从而诱导焦亡,而癌细胞表观遗传沉默对焦亡关键因子GSDME不表达,只发生了凋亡。如前所述,凋亡是一种“干净”的细胞死亡,如果化疗药物仅诱发凋亡,理论上不应产生很大的毒副作用。鉴于正常组织细胞表达GSDME而癌细胞基本不表达,邵峰实验室研究人员认为,GSDME介导的焦亡很可能是导致化疗药物产生毒副作用的原因。为了验证这个想法,研究者制备了GSDME敲除的小鼠,与正常小鼠用多种不同化疗药物进行对比实验,用化疗药物顺铂处理2周后的正常小鼠出现体重下降(下降幅度约为15%),肠道、脾脏和肺部组织均发生严重损伤和炎症反应,而在GSDME敲除小鼠上的器官组织损伤都有明显缓解。对于GSDME敲除的小鼠,化疗药物引起多种组织损伤和体重下降(下降幅度约为7%)都有明显的缓解,这些结果和细胞水平的数据也吻合,淋巴细胞和T细胞、B细胞下降速度减缓,很好地验证了研究者之前的想法。在其他几种化疗药物如5-氟尿嘧啶(5-FU)所致小鼠肠道损伤及博莱霉素所致肺部炎症模型上,结果也类似于顺铂处理的实验,并进一步验证了之前的结论。
邵峰院士表示,目前他正在思考两个重要问题,一是细胞焦亡在T细胞活化后会杀死肿瘤细胞的机制并不明确,GSDME被caspase-3切割活化,某些情况下靶细胞被T细胞杀伤,细胞焦亡也是参与的。一旦发生焦亡,炎症微环境就会发生变化。临床医师往往发现,进行化疗后的癌症患者再使用PD-1更有效,这是由于化疗后不可避免有细胞焦亡发生导致炎症微环境变化,巨噬细胞和T细胞的分化和功能都有变化。二是学术概念需要重新界定。多年来沿袭的概念是caspase-3诱导细胞凋亡,我们通过研究发现caspase-3诱导GSDME高表达也可以走细胞焦亡通路。“细胞死亡方式由底物决定,而不是由caspase决定。打个比方,caspase就像一把刀,如果用得恰当,为我们所用,杀掉入侵者可确保我们安全;如果用得不恰当,杀掉医生就没人看病了。”邵峰院士最后说。, http://www.100md.com
2015年12月7日,43岁的独立研究者邵峰当选中国科学院生命科学和医学学部院士,刷新了我国两院院士年龄榜,是1600多名院士中最年轻的一位。
1996年邵峰毕业于北京大学技术物理系应用化学专业,1999年中科院生物物理所硕士毕业,2003年获得美国密歇根大学医学院博士学位,2005年在哈佛大学医学院完成博士后训练后回国,在北京生命科学研究所创建实验室,带领团队开展独立研究生涯,在“细胞焦亡”领域做出了许多重要的开创性工作。回国前,邵峰在世界顶级学术期刊《细胞》(cell)和《科学》(science)杂志上分别发表了1篇论文。2005年回国后至今以通讯作者身份在《自然》(nature)、science、cell共发表论文11篇,研究成果多次获权威专家和学术杂志重点评述。目前邵峰实验室的研究继续保持着高水准,每一个成果都吸引了全球大量研究者的目光。
2013年,邵峰院士作为首位大陆本土科学家荣获国际蛋白质学会颁发的鄂文・西格青年科学家奖。2015年,他成为欧洲分子生物学组织(EMBO)外籍成员,此前只有5位中国科学家获选。他还曾先后荣获霍华德・休斯研究所青年科学家奖、吴阶平―保罗・杨森医学药学奖、周光召杰出青年基础科学奖和国家杰出青年基金。截至目前,邵峰实验室已发表学术论文70多篇,同行引用6000多次。
, http://www.100md.com
2017年,担任北京生命科学研究所(NIBS)学术副所长、资深研究员的邵峰院士带领团队首次揭示和阐明了细胞焦亡的机制。他们的研究结果显示,在细胞焦亡的发生机制中,半胱天冬酶(caspase)炎症小体下游的Gasdermin家族蛋白可能发挥了关键作用。
在2017年中国免疫学会年会上,邵峰院士深入浅出地阐述了细胞焦亡对人体免疫系统和癌症发病的相关机制。
他们在体外试验将小鼠骨髓诱导的巨噬细胞感染沙门氏杆菌(胞内菌)或痢疾杆菌(胞内自由生存的革兰氏阴性菌),同时将白细胞介素-1β(IL-1β)和染料加入培养基。感染后染料进入细胞核,免疫荧光显微镜观察到整个细胞发生爆炸性、裂解性死亡,这就是细胞焦亡(pyroptosis)。这一现象早在1992年就被观察到了,Nature杂志刊登了Zychlinsky A等的文章《弗氏志贺氏杆菌诱导感染的巨噬细胞凋亡》(Shigella flexneri induces apoptosis in infected macrophages),指出弗氏志贺氏杆菌可以诱导感染的宿主巨噬细胞发生程序性死亡。当时对程序性细胞死亡方式的认识仅限于凋亡,因此将其归结为细胞凋亡作用,这种认识是错误的。几年后的研究发现这种细胞死亡过程需要依赖半胱天冬酶-1(caspase-1)且伴随炎症发生,导致在2000年前后大家误将细胞凋亡和半胱氨酸蛋白酶 (caspase) 家族联系起来。但显然这一结论并不正确。这一细胞持续性死亡会发生细胞膜破裂,而细胞凋亡的细胞膜是不破裂的。 在20世纪90年代,人们对这种新的细胞死亡方式的性质不甚理解,并在较长时间内与细胞凋亡相混淆。
, 百拇医药
□邵峰教授在实验室
炎性caspase-1是怎么激活的?2002年瑞士Tschopp研究小组首次提出了炎症小体(Inflammasome)的概念,虽然没有提出实验证据。邵峰实验室的研究认为,炎症小体复合物能够激活胱冬肽酶-1(caspase-1),促进炎性细胞因子前体白介素-1(IL-1Lβ)和白介素-18(IL-18)切割成熟,同时激活的Caspase-1还可以使细胞发生焦亡。炎症小体在过去十几年里成为天然免疫领域最受关注的热点问题。各类炎症小体的区别在于不同的NLR 蛋白可以感受细胞内病原或危险信号的感染。其实当时没有实验证据存在炎症小体的复合物,更多是一种概念的提法,但确实有些疾病的证据支持这一说法,因为很多NLR家族基因的突变会导致一些自身炎症性疾病(AIDs)或自身免疫病。自身炎症性疾病主要是天然免疫系统发生问题,导致白介素-1(IL-1)活化。自身免疫病则是刺激人体免疫系统产生攻击自身的抗体。
邵峰院士介绍,我们实验室最早时探究NLR蛋白是怎么识别和清除细胞内的病原细菌。天然免疫反应的第一步是通过感知蛋白(受体蛋白)最先感知到细菌的入侵,此前对受体蛋白的研究主要集中在细胞膜上的一类天然免疫受体,因此,我们致力于发现宿主细胞质中感知细菌和病原细菌的感知蛋白(受体蛋白)的分子免疫机制,细菌内毒素(即脂多糖,LPS)被识别及其诱导炎症性坏死的机制是什么?我们创建了一种能将细菌内毒素(即脂多糖,LPS)高效导入哺乳动物细胞内的转染方法,发现LPS的胞内天然免疫受体是炎症性半胱天冬酶(caspase),并找到了全新的识别细菌主要模式分子的天然免疫受体(感知蛋白)NAIPs、Pyrin。其中,NAIPs蛋白可作为炎症小体的受体,识别细胞内细菌的鞭毛素蛋白和内毒素分泌系统的结构成分。Pyrin蛋白的主要功能是间接感知各种细菌毒力的作用,胞内的Rho家族小G蛋白修饰和失活导致肌动蛋白细胞骨架发生紊乱,进而活化经典炎症小体通路中的caspase-1。
, http://www.100md.com
20世纪90年代,Bruce Beutler等鉴定出Toll样受体4(TLR4)为细胞外的LPS的膜上受体,LPS结合TLR4后诱导细胞因子和炎症因子转录。邵峰由此产生了一个疑问:在细胞质里是否有受体蛋白来识别病原菌最为重要的模式分子LPS?两年前,邵峰实验室发现了一个炎症性Caspase家族,人细胞中的Caspase-4、Caspase-5是小鼠非经典炎症小体通路中Caspase-11的同源蛋白。与caspase-1类似的caspase-11可直接作为小鼠胞内受体识别革兰氏阴性菌的脂多糖(LPS),同时可诱导细胞焦亡的发生。
Toll样受体家族(NLRs)基因的突变还会导致自身炎症性疾病或自身免疫性疾病,这里面存在因果关系。比如NLR3基因突变可导致痛风、NLRP3突变可导致家族性寒冷性自身炎症综合征(FCAS)和穆一韦二氏综合征(MWS)、慢性婴儿神经皮肤关节综合征(CINCA)/NOMID(新生儿多系统炎症性疾病)、NLRC4激活的炎症小体突变可导致小肠结肠炎综合征和自身炎症(syndrome of enterocolitis and autoinflammation),也可导致自身免疫性疾病――复发性巨噬细胞活化综合征(recurrent macrophage activation syndrome)。MEFV基因编码的Pyrin功能获得性突变可导致自身炎症性疾病――家族性地中海热(FMF)。感知蛋白NAIPs、Pyrin的正常功能是什么?就是识别细菌的结构成分――脂多糖、鞭毛素蛋白。基因突变就是在未感染细菌时已被激活,从而导致各种疾病。这些炎症小体的基因突变会导致自身炎症性疾病,在自身免疫病发生中也起到辅助性作用。
, 百拇医药
细胞焦亡的“促成者”――GSDMD蛋白
细胞焦亡(pyroptosis)与细胞凋亡(apoptosis)究竟有什么差别?小鼠的巨噬细胞发生细胞凋亡时,细胞膜骤缩内陷裂成由细胞膜包裹的一个个泡状小体(即凋亡小体),凋亡小体表面有特殊信号导致其很快被巨噬细胞或邻近细胞识别并吞噬,不释放细胞内容物,因此不会导致组织水平的损伤和炎症反应,细胞的这种干净死亡(clean death)方式不影响其他细胞的正常功能。细胞焦亡是细胞体积不断胀大至细胞膜破裂,释放细胞内容物从而激活强烈的炎症反应,属于一种细胞炎性坏死(dirty death方式)。caspase-1/11都是在天然免疫的下游感知病原细菌的感染。caspase-1/11导致细胞焦亡发生的机制是什么?2015年,邵峰研究组运用全基因组编辑技术筛选人类基因组中的近2.2万个基因后,成功识别出一个新的基因――导致细胞焦亡的关键蛋白GSDMD并阐述了相关机制。GSDMD是所有炎性caspase的共有底物,其编码的蛋白质大约有500多个氨基酸和2个结构域氨基端gasdermin-N和羧基端gasdermin-C。正常状态下两个结构域相互“勾连”作用,处于无活性的自抑制状态,当炎性caspase-1或caspase-11活化后,可特异性切割GSDMD蛋白2个结构域之间的连接区域,释放的N端结构域可识别并结合细胞膜上的磷脂类分子,在细胞膜上打孔,导致细胞渗透压变化,细胞膜胀大裂解引发细胞焦亡。
, 百拇医药
细胞凋亡(左图)和细胞焦亡(右图)
由此可知,GSDMD蛋白与细胞焦亡密切相关,GSDMD蛋白是caspase-1和caspase-11/4/5诱导细胞焦亡的执行者,也清楚地阐述了细胞焦亡的本质是细胞炎性坏死。如果在细胞中去除GSDMD蛋白,细胞便不再发生细胞焦亡。长期以来人们都不甚清楚白细胞介素-1β(IL-1β)释放至细胞外的机制,邵峰团队的研究指出,GSDMD蛋白的缺失不会抑制激活caspase-1和对下游白介素1β的切割。在敲除GSDMD的细胞中,白细胞介素-1β可以被caspase-1切割成熟,但切割后成熟的白介素1β却几乎不能分泌到细胞外,显示细胞焦亡对于白介素1β的分泌必不可少,IL-1β不是通过分泌信号释放到细胞外,而是GSDMD在细胞膜上打孔后排出,GSDMD可能是IL-1β排出细胞外的必要条件。敲除GSDMD基因可以阻止白介素-1β的分泌和炎症小体及细菌LPS引起的细胞焦亡,使细胞转而发生凋亡。
caspase参与的炎症反应及细胞焦亡能有效提高机体抵抗内源性和外源性刺激的能力, 达到保护宿主的目的。caspase-1 和caspase-4/5/11介导的细胞焦亡通路具有天然免疫功能,当宿主细胞发生感染时可识别细菌鞭毛素蛋白、LPS,激活炎症小体介导的抗细菌天然免疫反应,当病原被天然免疫系统识别后启动焦亡。因此,将天然免疫受体基因敲除的小鼠无法清除感染的细菌而死亡,而正常野生型小鼠可以清除革兰氏阴性菌的感染而得以存活。实验中观察到,敲除GSDMD基因的小鼠脾脏和肝脏中,可以发现大量细菌的复制和病变。
, http://www.100md.com
另一方面,天然免疫系统也是双刃剑,细胞焦亡通路的过度活化也会引起负面反应。邵峰教授指出,革兰氏阴性菌导致的脓毒症是过度的细胞焦亡导致的。在小鼠模型注射大量LPS,它会在一天内死亡;如果把小鼠胞内受体caspase-11或下游的GSDMD敲除则可以存活。实验室还制作了一个抗体,只识别切割后的GSDMD蛋白,现在需要临床样本来验证这一抗体,有兴趣的临床医生可以与实验室取得联系。GSDMD蛋白是被caspase一分为二,一旦切割成功,细胞一定会发生凋亡。
邵峰实验室进一步研究发现,GSDMD代表了一个被称为gasdermin的家族性蛋白,除GSDMD外,GSDMA、GSDMB、GSDMC、GSDME(此 前 称DFNA5)、DFNB59等都具有N端和C端2个结构域,并能被炎性caspase切割。Gasdermin的家族蛋白中,仅GSDMD的2个结构域中间有caspase-1和-11的切割位点,但所有gasdermin家族性蛋白N端结构域被活化后都可在细胞膜上打孔从而诱导细胞焦亡。和GSDMD类似,未感染时这些蛋白N端和C端通过自抑制作用保持无活性状态。正因为此,邵峰团队最近将细胞焦亡重新定义为 “由Gasdermin家族蛋白介导的程序性细胞坏死”。由于GSDMD与炎症小体信号通路重要分子caspase-1和caspase-11/4/5相关,但其他Gasdermin家族蛋白与其无关,需要通过其他机制被切割活化,但最终也会通过启动细胞焦亡激活天然免疫反应。
, 百拇医药
邵峰实验室又在细胞焦亡研究领域取得了新的突破,揭示了另一种Gasdermin家族蛋白GSDME引起细胞焦亡的机制,研究结果已在线发表在2017年5月1日Nature杂志上。一组将实验用增殖表皮癌细胞(Hela Cells)中敲除GSDMD蛋白,回补GSDMD+肿瘤坏死因子a(TNFa),细胞遂发生了凋亡。另一组将Hela细胞的GSDMD敲除,回补一个改造过的可以被caspase-3切割活化的GSDMD(在两个结构域中间的caspase-1/4/11切割位点突变为caspase-3的切割位点,由于肿瘤坏死因子a诱导细胞凋亡是通过活化caspase-3),细胞从凋亡转化为焦亡,说明GSDMD蛋白只要在两个结构域中切割就会诱导细胞凋亡,这一发现对癌症治疗(尤其是化疗)的研发具有重要的指导意义。实验也说明细胞焦亡比细胞凋亡发生得更快。在第一组实验中可以观察到caspase-3切割活化的GSDMD,但另一组实验却观察不到caspase-3活化的GSDMD形态,这是由于焦亡发展得太快,掩盖了这一激活的过程,即细胞来不及凋亡就发生了焦亡。
, http://www.100md.com
邵峰实验室还完成了Gasdermin家族所有蛋白的对照实验。此前将GSDMD的caspase-1/4/11切割位点突变为caspase-3切割位点,可以中止TNFa诱导活化caspase-3引起的HeLa细胞凋亡,并转换为焦亡。在对照实验中,HeLa细胞中表达野生型的GSDME也同样可以将凋亡转换为焦亡,这是为什么呢?通过分析蛋白序列后发现,GSDME的N端和C端结构域中间存在天然的caspase-3切割位点。将caspase-3切割位点D突变为A,可以抑制caspase-3切割N端和C端结构域,细胞则恢复凋亡信号,不再发生焦亡。通过蛋白印迹实验(Westren Blot)中也证实,如果把caspase-3敲除或把切割位点D突变,就不能再切割N端和C端结构域,细胞焦亡也就不会发生。
另外一个有趣的现象是,肿瘤坏死因子a诱导细胞凋亡是通过活化caspase-3实现的,而caspase-3可以通过多种机制被活化。最经典的caspase-3活化机制是DNA损伤通过线粒体释放凋亡诱导因子,参与激活caspase-3。化疗药物顺铂等都会通过损伤DNA导致caspase-3活化。和GSDMD类似,caspase-3切割释放的GSDME的N端片段,能特异性结合4,5-二磷酸磷脂酰肌醇[PI(4,5)P2]并导致含有该磷脂的脂质体泄漏,负染电镜结果显示切割活化的GSDME可以在脂质体膜上打孔。这些结果说明GSDME是由caspase-3切割后活化,进而在膜上打孔并触发细胞焦亡。教科书上说caspase-3细胞凋亡,本质上是细胞炎性坏死。绝大多数常用的(癌)细胞系均不表达GSDME蛋白,所以被观察到的细胞确实是发生了凋亡。
, 百拇医药
邵峰实验室只找到了两株癌细胞表达高水平的内源GSDME――人的神经母细胞瘤细胞SH-SY5Y和恶性黑色素瘤细胞MeWo。把GSDME表达和不表达的细胞用化疗药物处理后,不表达GSDME的细胞用各种损伤DNA的化疗药物诱导后均发生了凋亡,而所有高表达GSDME的细胞都发生了焦亡。进一步检测了NCI-60(在培养基中生长的标准60种人类癌细胞系)的全部60株癌细胞后,邵峰实验室研究者发现,大多数的细胞不表达或低表达GSDME,仅有4~5个细胞表达较高水平的GSDME。查找现有文献发现,GSDME基因的启动子在癌化过程会发生DNA甲基化,导致表观遗传沉默。邵峰实验室于是做了一个很有意思的实验。不表达GSDME的癌细胞用地西他滨(目前唯一获准上市的DNA甲基化转移酶抑制剂地西他滨,可阻断DNA甲基化过程)处理后,成功诱导GSDME表达,此时再用传统化疗药物治疗,癌细胞对传统化疗药物变得更敏感(焦亡关键因子GSDME表达),更易发生炎性坏死。
很多临床医师开展的实验证明,地西他滨与传统化疗药物结合用于治疗骨髓增生异常综合征(MDS)效果更好,但找不到其中的机制。邵峰实验室的这项研究结果恰好解释了这一机制。小鼠的很多正常组织器官高表达GSDME,而癌化的细胞由于DNA甲基化介导的表观遗传沉默对焦亡关键因子GSDME不表达,使用地西他滨后激活caspase-3后诱导GSDME表达,从而促使癌细胞发生GSDME依赖的焦亡。
, 百拇医药
邵峰实验室进一步检测了5株人体正常组织来源的原代细胞血管内皮细胞,除2株不表达GSDME,另外3株高表达GSDME。经化疗药物处理这3株原代细胞后,GSDME高表达的细胞伴随caspase-3依赖的GSDME切割,发生焦亡;不表达GSDME的2株原代细胞则发生凋亡。在GSDME高表达的原代细胞中,用RNA干扰(RNAi)敲低GSDME表达后,化疗药物引起的细胞焦亡则转换为凋亡。
癌症患者使用化疗药物后为何会产生很大的毒副作用?因为在使用化疗药物后,正常原代细胞可高表达焦亡因子GSDME从而诱导焦亡,而癌细胞表观遗传沉默对焦亡关键因子GSDME不表达,只发生了凋亡。如前所述,凋亡是一种“干净”的细胞死亡,如果化疗药物仅诱发凋亡,理论上不应产生很大的毒副作用。鉴于正常组织细胞表达GSDME而癌细胞基本不表达,邵峰实验室研究人员认为,GSDME介导的焦亡很可能是导致化疗药物产生毒副作用的原因。为了验证这个想法,研究者制备了GSDME敲除的小鼠,与正常小鼠用多种不同化疗药物进行对比实验,用化疗药物顺铂处理2周后的正常小鼠出现体重下降(下降幅度约为15%),肠道、脾脏和肺部组织均发生严重损伤和炎症反应,而在GSDME敲除小鼠上的器官组织损伤都有明显缓解。对于GSDME敲除的小鼠,化疗药物引起多种组织损伤和体重下降(下降幅度约为7%)都有明显的缓解,这些结果和细胞水平的数据也吻合,淋巴细胞和T细胞、B细胞下降速度减缓,很好地验证了研究者之前的想法。在其他几种化疗药物如5-氟尿嘧啶(5-FU)所致小鼠肠道损伤及博莱霉素所致肺部炎症模型上,结果也类似于顺铂处理的实验,并进一步验证了之前的结论。
邵峰院士表示,目前他正在思考两个重要问题,一是细胞焦亡在T细胞活化后会杀死肿瘤细胞的机制并不明确,GSDME被caspase-3切割活化,某些情况下靶细胞被T细胞杀伤,细胞焦亡也是参与的。一旦发生焦亡,炎症微环境就会发生变化。临床医师往往发现,进行化疗后的癌症患者再使用PD-1更有效,这是由于化疗后不可避免有细胞焦亡发生导致炎症微环境变化,巨噬细胞和T细胞的分化和功能都有变化。二是学术概念需要重新界定。多年来沿袭的概念是caspase-3诱导细胞凋亡,我们通过研究发现caspase-3诱导GSDME高表达也可以走细胞焦亡通路。“细胞死亡方式由底物决定,而不是由caspase决定。打个比方,caspase就像一把刀,如果用得恰当,为我们所用,杀掉入侵者可确保我们安全;如果用得不恰当,杀掉医生就没人看病了。”邵峰院士最后说。, http://www.100md.com